
APOBEC Mutagenesis in Cancer Development and Susceptibility
1
Molecular Biology Program, Sloan Kettering Institute, Memorial Sloan Kettering Cancer Center, New York, NY 10065, USA
2
Medical Scientist Training Program, New York University Grossman School of Medicine, New York, NY 10016, USA
3
Department of Pathology, NYU Grossman School of Medicine, New York, NY 10016, USA
4
Perlmutter Cancer Center, NYU Grossman School of Medicine, New York, NY 10016, USA
*
Author to whom correspondence should be addressed.
Cancers 2024, 16(2), 374; https://doi.org/10.3390/cancers16020374
Submission received: 20 December 2023
/
Revised: 9 January 2024
/
Accepted: 11 January 2024
/
Published: 15 January 2024
(This article belongs to the Special Issue The Study of Cancer Susceptibility Genes (Volume II))
Abstract
:Simple Summary
APOBEC cytosine deaminases represent potent mutational sources in over 50% of human cancers and are linked to tumor heterogeneity and therapy responses. However, the understanding of the contribution of APOBEC-mediated mutagenesis to cancer susceptibility and malignant transformation is still limited. The authors review the existing evidence for the impact of APOBEC mutagenesis on cancer development and identify gaps in related knowledge that need to be addressed.
Abstract
APOBEC cytosine deaminases are prominent mutators in cancer, mediating mutations in over 50% of cancers. APOBEC mutagenesis has been linked to tumor heterogeneity, persistent cell evolution, and therapy responses. While emerging evidence supports the impact of APOBEC mutagenesis on cancer progression, the understanding of its contribution to cancer susceptibility and malignant transformation is limited. We examine the existing evidence for the role of APOBEC mutagenesis in carcinogenesis on the basis of the reported associations between germline polymorphisms in genes encoding APOBEC enzymes and cancer risk, insights into APOBEC activities from sequencing efforts of both malignant and non-malignant human tissues, and in vivo studies. We discuss key knowledge gaps and highlight possible ways to gain a deeper understanding of the contribution of APOBEC mutagenesis to cancer development.
1. Introduction
The AID (activation-induced cytidine deaminase)/APOBEC (apolipoprotein B mRNA editing enzyme catalytic subunit) family comprises eleven members as follows: AID, APOBEC1, APOBEC2, APOBEC3A, APOBEC3B, APOBEC3C, APOBEC3D/E, APOBEC3F, APOBEC3G, APOBEC3H, and APOBEC4. While APOBEC2 and APOBEC4 members lack known deaminase activities, other enzymes play pivotal roles in immune and metabolic processes [1,2,3,4,5,6,7]. Put briefly, AID-mediated cytosine deamination at immunoglobulin loci contributes to somatic hypermutation and antibody diversification; APOBEC1-mediated cytidine deamination generates a lower molecular weight form of apolipoprotein B (ApoB) in the small intestine, which is essential for triglyceride transport; and the APOBEC3 subfamily-mediated deamination of retroviral and viral cytosines and cytidines limits viral replication as part of an innate immune defense [1,2,3,4,5,6,7].
Certain AID/APOBEC enzymes emerged as prominent mutators in cancer. APOBEC1 and several APOBEC3 members (3A, 3B, 3C, 3D/E, 3F, 3H) preferentially deaminate cytosine bases in TC dinucleotides, which can lead to mutations in targeted cytosines [8,9,10,11,12,13]. Mutational signatures that are characterized by cytosine mutations in TC dinucleotides, which are reflective of APOBEC1 and relevant APOBEC3 activities in cancer genomes, have been detected in over 50% of cancers and most cancer types [14,15]. These include the single-base substitution (SBS) signatures of genome-wide non-clustered C > T (called signature “SBS2”) and C > G/A (SBS13) mutations in TC dinucleotides [14] as well as the signatures of clustered cytosine mutations in TC dinucleotides, kataegis (local strand-coordinated hypermutation) [15] and omikli (diffuse hypermutation) [16]. APOBEC3A and APOBEC3B are the only endogenous enzymes that are confirmed to induce these signatures in human cells [17,18], with indications that additional APOBEC deaminases may contribute to cancer mutagenesis [11,19,20]. Other mutational types linked to direct or indirect APOBEC activities include APOBEC3A-mediated small insertions, deletions [21] as well as substitutions in non-TC dinucleotide cytosines in certain palindromic sequences [22], APOBEC3G-mediated SBS mutations [20], a doublet base substitution signature [14], and structural as well as copy number variations [9,23,24,25]. Mutations associated with AID activities are also found in cancer genomes. AID-mediated cytosine deamination during somatic hypermutation directly induces mutations in C:G pairs in WRC (W = A or T base; R = A or G) motifs and indirectly contributes to mutations in T:A pairs [7]. Clustered mutations linked to direct and indirect AID activities (respectively, SBS84 and SBS85 in census COSMIC database signatures [14]) are frequently detected in immunoglobulin heavy chain variable region (IGHV) genes in chronic myeloid leukemia (CLL), multiple myeloma, and diffuse large B-cell lymphomas (DLBCLs) [26,27,28,29,30]. Although a non-clustered genome-wide signature (SBS9) was initially proposed to be associated with AID activity [31], recent data suggest otherwise [32]. AID activity primarily targets IGHV, but it can also affect other regions with a preferential targeting of ±2 kb from the transcription start sites of highly transcribed genes [26]. Mutations linked to off-target AID activities are generally higher in cases with a mutated IGHV [26,27,33]. Additionally, AID can also induce rearrangements that are frequently found in implicated cancer types [34].
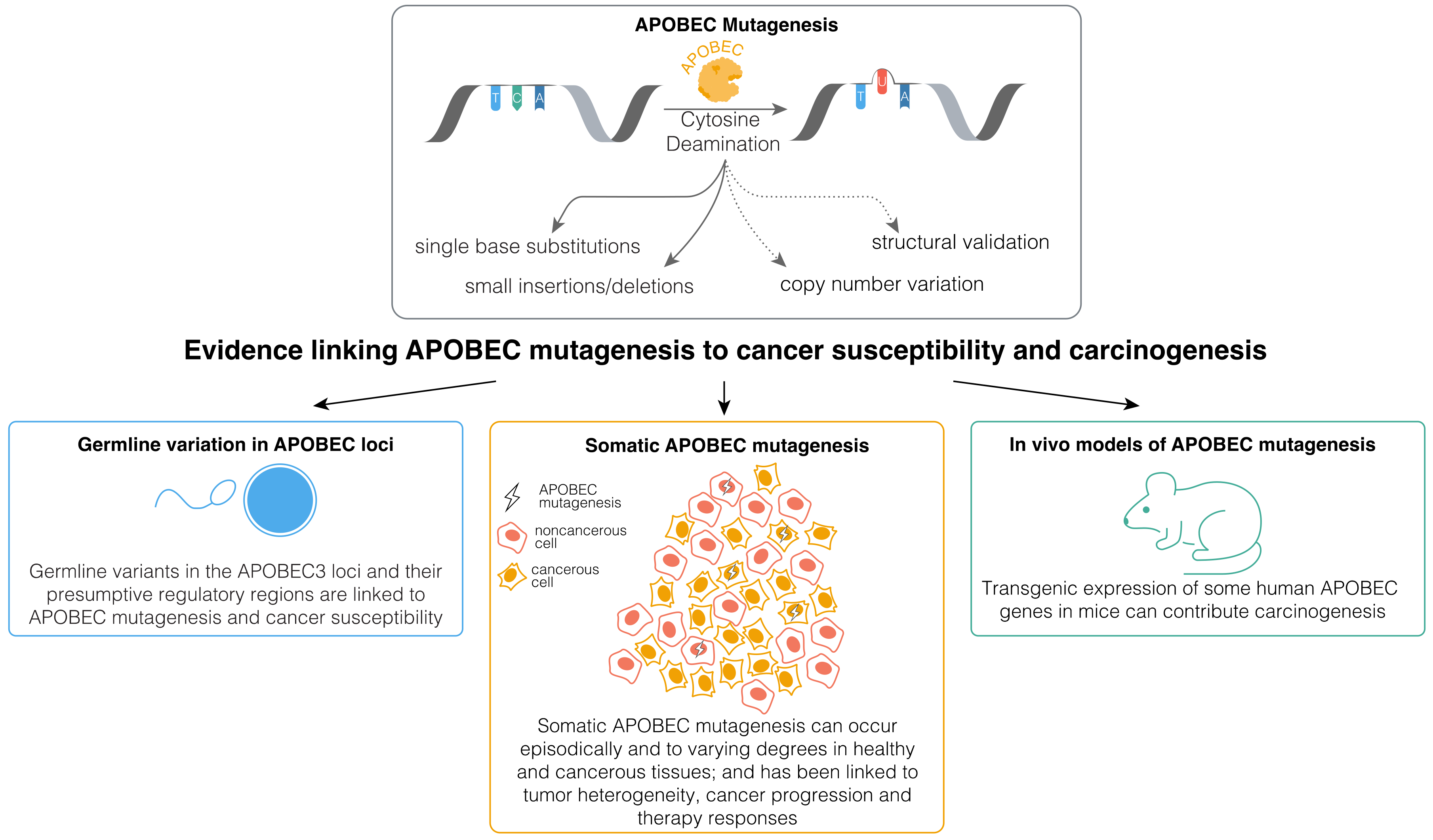
AID has been implicated in cancer development and progression, with the related roles extensively reviewed recently [35]. However, the precise contributions of APOBEC deaminases to cancer evolution remains less well understood. While APOBEC enzymes may contribute to cancer evolution through non-mutagenic mechanisms [25,36,37,38,39,40], mutagenesis by these enzymes appears to have a more widespread impact on cancer [20,36,41,42,43,44,45,46]. APOBEC mutagenesis endures in vitro in human cancer cell lines [47], and its signatures often appear in the subclonal phylogenetic branches of primary tumors and metastatic cancers, with incidental observations of driver mutations in APOBEC-associated sequence contexts [48,49,50,51,52,53,54]. APOBEC3A and APOBEC3B have been linked to persistent cell evolution and therapy resistance in lung cancers [43,45,46], and APOBEC3B has been linked to resistance against androgen receptor (AR)-targeted therapy and Tamoxifen in prostate and estrogen receptor-positive (ER+) breast cancers [42,55]. Furthermore, in vivo studies suggest that APOBEC mutagenesis can promote tumor heterogeneity [20,56,57,58]. These data and others indicate that ongoing APOBEC mutagenesis likely plays a significant role in cancer progression, although further experimental validation is necessary, as we have discussed before [41]. However, the contribution of APOBEC mutagenesis to malignant transformation remains considerably less well understood. Here, we outline the existing evidence (Figure 1) for the role of APOBEC mutagenesis in carcinogenesis and cancer susceptibility, addressing key knowledge gaps and discussing possible ways forward in order to address them.
2. Germline Variants Implicating APOBEC Mutagenesis in Cancer Susceptibility
Several polymorphisms in genes encoding APOBEC enzymes have been associated with a differential risk of cancer. One such polymorphism, a 29.5 kb deletion of the consecutive 3′-end of the APOBEC3A and most of the APOBEC3B gene found on chromosome 22 (A3AB deletion), produces a hybrid sequence of APOBEC3A fused with the 3′-untranslated region (UTR) of APOBEC3B [59]. The prevalence of A3AB deletion varies across ethnicities (Southeast Asian, 36.9%; South American, 57.7%; African, 0.9%; European, 6%) [59] and has been associated with the increased risks of breast and ovarian cancers among Asian populations [60,61,62,63]. The links between the A3AB deletion and cancer risk in Europeans are conflicting [64,65,66,67], although carriers under 50 years of age show strong indications of an increased risk in lung and prostate cancers [66]. The mechanisms underpinning the links between the A3AB deletion and cancer risk are not well understood. Proposed explanations include the stabilized expression of the hybrid APOBEC3A’s transcript [68] and the increased nuclear localization of APOBEC3H conferred by the A3H-I haplotype found in relevant polymorphism carriers [11], both of which are predicted to generate more potent mutator enzymes. Indeed, breast cancers from carriers exhibit elevated mutational burdens of APOBEC-mediated SBS2 and SBS13 signatures, with more mutations in homozygous carriers compared with heterozygous ones [69,70]. Thus, the available data imply that polymorphism may confer an increased risk of cancer as a result of the overactivity of certain APOBEC enzyme(s) and a consequential increase in mutational burdens.
Another polymorphism in the APOBEC-related locus, a single nucleotide polymorphism (SNP) rs1014971 (allele: T), has been associated with an increased risk of bladder cancer [71,72,73], APOBEC3B expression [71], and APOBEC-signature mutational burdens in bladder tumors [71]. This SNP is located in a putative long-distance enhancer region upstream from the APOBEC3 cluster that can interact with the APOBEC3B promoter [74], potentially leading to an elevated APOBEC3B expression and APOBEC-associated mutational burdens [71]. Rs1014971 has also been associated with an increased cancer risk and the APOBEC3B expression in breast cancer [71]. However, breast cancers from polymorphism carriers do not display an increased number of APOBEC-mediated mutations [71]. It is possible that the increased expression of APOBEC3B associated with rs1014971 may contribute to breast cancer susceptibility through mutagenesis-independent mechanisms, some of which have been proposed’ as mechanisms with such a function before [40].
Overall, the link between germline polymorphisms in APOBEC loci and cancer risk strongly implicate APOBEC enzymes in cancer susceptibility. Experimental work is required to validate existing associations and to understand the underpinning mechanisms. Mechanistic insights combined with genome-wide association studies across larger and broader populations will be critical for understanding the differences in risks across populations and the cancer types conferred by polymorphisms in APOBEC-related loci.
3. Somatic Mutagenesis Implicating APOBEC Mutagenesis in Cancer Susceptibility
The hypothesis that APOBEC mutagenesis plays a role in carcinogenesis assumes that it induces driver mutations that contribute to malignant transformations. Somatic APOBEC-associated mutational signatures and driver mutations have been detected in many types of primary cancers [14,75]. However, it is often unclear whether the relevant driver mutations occur during or after a malignant transformation. Advances in DNA sequencing strategies [76] have enabled insights into APOBEC mutagenesis in non-malignant and pre-malignant human tissues, providing a glimpse into its activities before a malignant transformation is complete (summarized in Table 1). These emerging insights combined with data from cancer genome sequences are key to understanding the potential of APOBEC mutagenesis to contribute to associated cancer types.
The APOBEC-mediated SBS2 and SBS13 are either very rare or absent in hepatocellular carcinoma, testicular cancer, thyroid adenocarcinoma [14,96], and their respective non-malignant tissue types [78,79,91], implying that APOBEC mutagenesis may not play a significant role in the development of these cancer types. On the other hand, SBS2 and SBS13 are found in the majority of esophageal squamous cell carcinomas (ESCCs) but are rare in a normal esophageal epithelium, where they are detected in ~0–<5% individuals [14,97,98]. A study on ESCC development found that these signatures are also rare (detected in ~4% of clones) and contribute low mutational burdens in cases of low-grade intraepithelial neoplasia (LGIN), with hypermutation only detected in cases of high-grade intraepithelial neoplasia (HGIN) where the relevant signatures presented themselves in ~25% of clones [85]. Nevertheless, both LGIN and HGIN clones harboring the APOBEC-mediated mutations exhibited TP53 biallelic loss and high levels of copy number alterations. These data suggest that APOBEC hypermutation occurs after acquiring initial TP53 mutations and is likely to not be a major contributor to genome instability in the early stages of ESCC [85,97]. Indeed, APOBEC-mediated mutational signatures are absent from the spectrum of TP53 mutations in ESCC [97]. Similarly, APOBEC-mediated signatures have been identified in pancreatic adenocarcinomas (~46%), endometrial adenocarcinomas (~11%), acute lymphoblastic leukemia (~11%), B-cell lymphomas (~10%), stomach adenocarcinomas (~19%), renal-cell carcinomas (~18%), liposarcomas (~95%), adrenocortical cancers (~71%) and biliary adenocarcinomas (48%) [14,97,99]; however, they have not been identified in cells from respective healthy tissue types [32,78,79,92,93]. It is possible that APOBEC mutagenesis becomes active only during the later stages of cancer development, as observed in the esophagus. In contrast, APOBEC-mediated mutations occur to varying degrees in the bronchial epithelium of a healthy lung [77,78] (detected in 11–78% of samples depending on the study), small [19,78,79] (14–73%) as well as colonic [82] (0.5%) intestinal crypts, bladder urothelium [80] (22%), and in cancers from corresponding tissue types, which are sometimes according to similar proportions [14,19,100]. APOBEC mutagenesis may thus play a more relevant role in the early stages of the development of these cancers.
Overall, available data indicate that APOBEC mutagenesis can occur in non-malignant tissues, albeit with varying rates of prevalence across different tissue types, in a similar way to cancers [14]. Therefore, the potential contribution of APOBEC mutagenesis to carcinogenesis likely differs across tissue types due to the variable mechanisms and extents of APOBEC regulation and/or dysregulation. Future studies examining APOBEC-mediated mutational signatures and driver mutations across different stages of cancer cell evolution from larger cohorts are critical to determining the timing of such events and the extent to which APOBEC mutagenesis may contribute to the evolution of individual cancer types. Such studies should also encompass other tissues where APOBEC mutagenesis is prevalent in cancer, such as breast and ovary. Additionally, APOBEC mutagenesis is prevalent in cervical cancers as well as in head and neck squamous cell carcinomas (HNSCCs) where it has been proposed to contribute to cancer development as a consequence of misdirected activity against human papillomavirus (HPV), which is associated with the etiologies of these cancers [31,101]. Comparative analyses of samples affected and unaffected by the relevant viruses can provide further insight into the role of APOBEC deaminases in the etiology of HPV-associated cancer types. For example, APOBEC-mediated mutations are more common in HPV-positive HNSCCs where they have been linked to the generation of oncogenic PIK3CA mutations, unlike in HPV-negative HNSCCs [101]. When premalignant tissue samples are not routinely biopsied, such as those of HPV-positive HNSCC cancers, computational models that infer the phylogenetic relationship between tumor subclones can be utilized to predict the timing of the somatic events in carcinogenesis [102].
4. In Vivo Data Implicating APOBEC Mutagenesis in Carcinogenesis
In vivo studies (summarized in Table 2) have offered valuable insight into the potential of APOBEC mutagenesis to contribute to cancer development. An early study found that transgenic mice and rabbits expressing rabbit APOBEC1 in their livers developed hepatocellular carcinomas, while controls did not [103]. However, it is not clear whether APOBEC1 contributes to carcinogenesis through mutagenesis or an alternative mechanism. Importantly, the impact of APOBEC1 on human tissues is uncertain as it is typically expressed in the small intestine and duodenum but not the liver [19]. Additionally, APOBEC-mediated signatures have not been detected or do not contribute to many mutations in respective cancer types [14,96,100,104]. In vivo examination of the roles of APOBEC3 mutagenesis has largely relied on transgenic models due to the lack of most human APOBEC3 ortholog genes in mice [105]. In models predisposed to colorectal cancer (ApcMin), the constitutive, ubiquitous expression of human tumor-like levels of APOBEC3A from the CAG promoter can promote the development of colorectal cancer [57]. Similarly, higher, likely transient, levels of APOBEC3A through an integrative transposable element can elevate liver cancer rates in Fah liver regeneration models, which are contingent on APOBEC3A catalytic activity [57,58]. In both models, induction of APOBEC3A inflicted APOBEC-associated mutations. Interestingly, expression of the other six APOBEC3 paralogs, including APOBEC3B, failed to develop tumors in the Fah models [57]. However, a subsequent study found that the constitutive, ubiquitous expression of higher and human tumor-like APOBEC3B levels from CAG the promoter accelerates liver and lymphoma cancer formation as well as metastasis in non-predisposed animals [56]. Tumors from APOBEC3B-expressing mice accumulated a higher proportion of APOBEC-mediated SBS2 mutations, with all APOBEC3B-related phenotypes dependent on its catalytic activity, although the total mutational burdens in developed tumors were similar to those in controls.
Collectively, these findings demonstrate that APOBEC3A- and APOBEC3B-mediated deamination can promote carcinogenesis. However, it remains less defined how well the relevant models reflect enzyme activities in human tissues. Inducing APOBEC3A and APOBEC3B for tumor levels may not accurately mimic the carcinogenesis arising in pre-malignant tissues given their generally lower expression in normal tissues [106,107,108]. Additionally, APOBEC mutagenesis can occur transiently in both human cancer cell lines [47] and non-malignant tissues [19,77] where it can, similarly to human cancers, only affect certain cellular lineages [19,52,77,80,81,82,109] and be infrequent over their lifetimes [19]. The constitutive, ubiquitous expression of APOBEC enzymes from a commonly used heterologous CAG promoter does not recapitulate these features since it separates expression from the regulatory mechanisms in human cells [56]. Indeed, depending on the level, duration, and model system, the expression of APOBEC3B can lead to various outcomes, including that of there being no overt tumor phenotypes [57,110], increased tumor rates [56], detriments to tumor development [45], or lethality [111]. This variability underscores the importance of inducing APOBEC enzymes under conditions that accurately replicate relevant human tissue settings.

{kind=link}
Table 2.
Existing APOBEC transgenic in vivo models. The “Mouse Model” column details the mouse strain upon which APOBEC induction was performed. The “APOBEC Induction Strategy” column outlines the specific APOBEC gene induced, localization, and timing of induction. The “Level of Induction” column indicates the level of APOBEC induction, while the ‘Phenotype’ column summarizes the resulting characteristics or effects upon induction.
Table 2.
Existing APOBEC transgenic in vivo models. The “Mouse Model” column details the mouse strain upon which APOBEC induction was performed. The “APOBEC Induction Strategy” column outlines the specific APOBEC gene induced, localization, and timing of induction. The “Level of Induction” column indicates the level of APOBEC induction, while the ‘Phenotype’ column summarizes the resulting characteristics or effects upon induction.
| Study | Mouse Model | APOBEC Induction Strategy (Gene, Localization, Induction) | Level of Induction | Phenotype |
|---|---|---|---|---|
| Yamanaka et al., 1995 [103]. PMID: 7667315 | Wild-type (no cancer predisposition) | APOBEC1, ectopic (liver), stable | Overexpressed | APOBEC1 expression causes the development of liver dysplasia and hepatocellular carcinomas. Transgenic animals contain transcripts that are extensively edited by APOBEC1. |
| Law et al., 2020 [57]. PMID: 32870257 | Wild-type (no cancer predisposition) | APOBEC3A, ubiquitous, constitutive | Human tumor-like levels | APOBEC3A expression is insufficient for tumor initiation. |
| Boumelha et al., 2022 [110]. PMID: 35930804 | Wild-type (no cancer predisposition) | APOBEC3B, ubiquitous, Cre-induced | Not reported | APOBEC3B expression does not induce tumors. |
| Boumelha et al., 2022 [110]. PMID: 35930804 | KrasLSL-G12D/+, Trp53fl/fl-driven lung cancer model | APOBEC3B, ubiquitous, Cre-induced | Not reported | APOBEC3B expression does not increase tumor growth rate and fails to substantially increase clonal tumor mutational burden. |
| Boumelha et al., 2022 [110]. PMID: 35930804 | Urethane-induced lung cancer model | APOBEC3B, ubiquitous, Cre-induced | Not reported | APOBEC3B expression does not increase tumor growth rate or the number of tumors per animal. |
| Law et al., 2020 [57]. PMID: 32870257 | Adenomatous polyposis coli multiple intestinal neoplasia (Apcmin)-driven colon cancer model | APOBEC3A, ubiquitous, constitutive | Human tumor-like levels | APOBEC3A expression in murine colon tissue increases tumorigenesis and APOBEC-associated mutations. |
| Law et al., 2020 [57]. PMID: 32870257 | Adenomatous polyposis coli multiple intestinal neoplasia (Apcmin)-driven colon cancer model | APOBEC3G, ubiquitous, constitutive | Not reported | APOBEC3G expression does not increase polyp formation. |
| Law et al., 2020 [57]. PMID: 32870257 | Fumaryl-acetoacetate hydrolase (Fah) model for hepatocellular carcinoma (with shTp53) | APOBEC3(A-H), hydrodynamic transfer at 2 months | Not reported | APOBEC3A expression in murine liver tissue increases tumorigenesis and APOBEC mutations (SBS2 and SBS13). Other APOBEC3 paralogs fail to develop tumors. |
| Naumann et al., 2023 [58]. PMID: 37298259 | Fumaryl-acetoacetate hydrolase (Fah) model for hepatocellular carcinoma | APOBEC3A, hydrodynamic transfer at 2 months | Not reported | APOBEC3A is capable of driving tumor development. Catalytic activity and DNA deamination (not RNA-editing) are required to promote tumor formation. |
| Durfee et al., 2023 [56]. PMID: 37797615 | Wild-type (no cancer predisposition) | APOBEC3B, ubiquitous, constitutive | Human tumor-like levels | APOBEC3B expression accelerates rates of carcinogenesis, tumor heterogeneity, and metastasis in older animals. Transgenic animals display an increase in APOBEC-associated mutations, indels, and structural variations. APOBEC3B catalytic activity is required for all phenotypes. |
| Caswell et al., 2023 [45]. PMID: 38049664 | Tp53 WT, EGFRL858R-driven lung cancer model | APOBEC3B, ubiquitous, induced at tumor initiation | Not reported | APOBEC3B expression constrains tumorigenesis. Catalytic activity is required for the phenotype. |
| Liu et al., 2023 [20]. PMID: 36480186 | n-butyl-N-(4-hydroxybutyl)nitrosamine (BBN)-induced bladder cancer model | APOBEC3G, ubiquitous, constitutive | Not reported | APOBEC3G expression promotes mutagenesis, genomic instability, and kataegis, leading to shorter survival in animals. A novel SBS signature is identified in animals expressing APOBEC3G. |
| Wormann et al., 2021 [25]. PMID: 35121902 | Pdx1-Cre, KRASG12D, Tp53fl/fl-driven pancreatic cancer model | APOBEC3A (truncated), ubiquitous, stable | Similar to A3A levels in human lymphocytes (physiological levels of A3A compared with human ones) | APOBEC3A expression leads to more aggressive tumors and metastasis independent of its canonical deaminase functions. |
| de la Vega et al., 2023 [111]. PMID: 38001542 | Wild-type (no cancer predisposition) | APOBEC3B, ubiquitous, induced at 4 weeks s | In lungs, expression is within the range observed in human cancers. In the liver and pancreas, expression is comparable to human tumors with highest APOBEC3B levels and is associated with poor survival. | APOBEC3B expression leads to RNA editing and is lethal. |
5. Conclusions
Overall, murine studies and insights from human genome sequencing efforts indicate that APOBEC enzymes can contribute to carcinogenesis, but further research is necessary to comprehend the mechanisms and extent of APOBEC mutagenetic involvement in the development of different human cancer types. Examination of APOBEC-associated mutational signatures and driver mutations across pre-malignant tissues and tissues reflective of the transitional stages during carcinogenesis, as well as development of models that closely resemble mutagenic APOBEC activities emerging from such studies, will be critical to assess their contribution to the development of different human cancer types.
Author Contributions
Conceptualization: M.P.; Literature Review: A.D., J.S., J.S.R. and M.P.; Writing—Original Draft Preparation: A.D., J.S., J.S.R. and M.P.; Writing—Review and Editing: A.D., J.S., J.S.R. and M.P.; Visualization: J.S.; Supervision: M.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Institutes of Health grant number CA270102.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Conflicts of Interest
M.P. is a shareholder in Vertex Pharmaceuticals and a compensated consultant for the GLG Network.
References
- Conticello, S.G. The AID/APOBEC family of nucleic acid mutators. Genome Biol. 2008, 9, 229. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Dudley, J.P. APOBECs and virus restriction. Virology 2015, 479–480, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Smith, H.C.; Bennett, R.P.; Kizilyer, A.; McDougall, W.M.; Prohaska, K.M. Functions and regulation of the APOBEC family of proteins. Semin. Cell Dev. Biol. 2012, 23, 258–268. [Google Scholar] [CrossRef]
- Marino, D.; Perković, M.; Hain, A.; Jaguva Vasudevan, A.A.; Hofmann, H.; Hanschmann, K.M.; Mühlebach, M.D.; Schumann, G.G.; König, R.; Cichutek, K.; et al. APOBEC4 Enhances the Replication of HIV-1. PLoS ONE 2016, 11, e0155422. [Google Scholar] [CrossRef] [PubMed]
- Lada, A.G.; Krick, C.F.; Kozmin, S.G.; Mayorov, V.I.; Karpova, T.S.; Rogozin, I.B.; Pavlov, Y.I. Mutator effects and mutation signatures of editing deaminases produced in bacteria and yeast. Biochemistry 2011, 76, 131–146. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, J.; Evans, T.; Kumar, R.; DiMenna, L. Biological function of activation-induced cytidine deaminase (AID). Biomed. J. 2014, 37, 269–283. [Google Scholar] [CrossRef]
- Di Noia, J.M.; Neuberger, M.S. Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem. 2007, 76, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.S.; Petersen-Mahrt, S.K.; Neuberger, M.S. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell 2002, 10, 1247–1253. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.J.; Nik-Zainal, S.; Wu, Y.L.; Stebbings, L.A.; Raine, K.; Campbell, P.J.; Rada, C.; Stratton, M.R.; Neuberger, M.S. DNA deaminases induce break-associated mutation showers with implication of APOBEC3B and 3A in breast cancer kataegis. elife 2013, 2, e00534. [Google Scholar] [CrossRef] [PubMed]
- Suspène, R.; Aynaud, M.-M.; Guétard, D.; Henry, M.; Eckhoff, G.; Marchio, A.; Pineau, P.; Dejean, A.; Vartanian, J.-P.; Wain-Hobson, S. Somatic hypermutation of human mitochondrial and nuclear DNA by APOBEC3 cytidine deaminases, a pathway for DNA catabolism. Proc. Natl. Acad. Sci. USA 2011, 108, 4858–4863. [Google Scholar] [CrossRef] [PubMed]
- Starrett, G.J.; Luengas, E.M.; McCann, J.L.; Ebrahimi, D.; Temiz, N.A.; Love, R.P.; Feng, Y.; Adolph, M.B.; Chelico, L.; Law, E.K.; et al. The DNA cytosine deaminase APOBEC3H haplotype I likely contributes to breast and lung cancer mutagenesis. Nat. Commun. 2016, 7, 12918. [Google Scholar] [CrossRef]
- Bishop, K.N.; Holmes, R.K.; Sheehy, A.M.; Davidson, N.O.; Cho, S.-J.; Malim, M.H. Cytidine deamination of retroviral DNA by diverse APOBEC proteins. Curr. Biol. 2004, 14, 1392–1396. [Google Scholar] [CrossRef] [PubMed]
- Dang, Y.; Wang, X.; Esselman, W.J.; Zheng, Y.-H. Identification of APOBEC3DE as another antiretroviral factor from the human APOBEC family. J. Virol. 2006, 80, 10522–10533. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Ng, A.W.T.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Alexandrov, L.B.; Wedge, D.C.; Van Loo, P.; Greenman, C.D.; Raine, K.; Jones, D.; Hinton, J.; Marshall, J.; Stebbings, L.A.; et al. Mutational processes molding the genomes of 21 breast cancers. Cell 2012, 149, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Mas-Ponte, D.; Supek, F. DNA mismatch repair promotes APOBEC3-mediated diffuse hypermutation in human cancers. Nat. Genet. 2020, 52, 958–968. [Google Scholar] [CrossRef]
- Petljak, M.; Dananberg, A.; Chu, K.; Bergstrom, E.N.; Striepen, J.; von Morgen, P.; Chen, Y.; Shah, H.; Sale, J.E.; Alexandrov, L.B.; et al. Mechanisms of APOBEC3 mutagenesis in human cancer cells. Nature 2022, 607, 799–807. [Google Scholar] [CrossRef]
- Maciejowski, J.; Chatzipli, A.; Dananberg, A.; Chu, K.; Toufektchan, E.; Klimczak, L.J.; Gordenin, D.A.; Campbell, P.J.; de Lange, T. APOBEC3-dependent kataegis and TREX1-driven chromothripsis during telomere crisis. Nat. Genet. 2020, 52, 884–890. [Google Scholar] [CrossRef]
- Wang, Y.; Robinson, P.S.; Coorens, T.H.H.; Moore, L.; Lee-Six, H.; Noorani, A.; Sanders, M.A.; Jung, H.; Katainen, R.; Heuschkel, R.; et al. APOBEC mutagenesis is a common process in normal human small intestine. Nat. Genet. 2023, 55, 246–254. [Google Scholar] [CrossRef]
- Liu, W.; Newhall, K.P.; Khani, F.; Barlow, L.; Nguyen, D.; Gu, L.; Eng, K.; Bhinder, B.; Uppal, M.; Récapet, C.; et al. The Cytidine Deaminase APOBEC3G Contributes to Cancer Mutagenesis and Clonal Evolution in Bladder Cancer. Cancer Res. 2023, 83, 506–520. [Google Scholar] [CrossRef]
- DeWeerd, R.A.; Németh, E.; Póti, Á.; Petryk, N.; Chen, C.-L.; Hyrien, O.; Szüts, D.; Green, A.M. Prospectively defined patterns of APOBEC3A mutagenesis are prevalent in human cancers. Cell Rep. 2022, 38, 110555. [Google Scholar] [CrossRef] [PubMed]
- Langenbucher, A.; Bowen, D.; Sakhtemani, R.; Bournique, E.; Wise, J.F.; Zou, L.; Bhagwat, A.S.; Buisson, R.; Lawrence, M.S. An extended APOBEC3A mutation signature in cancer. Nat. Commun. 2021, 12, 1602. [Google Scholar] [CrossRef] [PubMed]
- Jakobsdottir, G.M.; Brewer, D.S.; Cooper, C.; Green, C.; Wedge, D.C. APOBEC3 mutational signatures are associated with extensive and diverse genomic instability across multiple tumour types. BMC Biol. 2022, 20, 117. [Google Scholar] [CrossRef]
- Venkatesan, S.; Angelova, M.; Puttick, C.; Zhai, H.; Caswell, D.R.; Lu, W.-T.; Dietzen, M.; Galanos, P.; Evangelou, K.; Bellelli, R.; et al. Induction of APOBEC3 Exacerbates DNA Replication Stress and Chromosomal Instability in Early Breast and Lung Cancer Evolution. Cancer Discov. 2021, 11, 2456–2473. [Google Scholar] [CrossRef] [PubMed]
- Wörmann, S.M.; Zhang, A.; Thege, F.I.; Cowan, R.W.; Rupani, D.N.; Wang, R.; Manning, S.L.; Gates, C.; Wu, W.; Levin-Klein, R.; et al. APOBEC3A drives deaminase domain-independent chromosomal instability to promote pancreatic cancer metastasis. Nat. Cancer 2021, 2, 1338–1356. [Google Scholar] [CrossRef] [PubMed]
- Kasar, S.; Kim, J.; Improgo, R.; Tiao, G.; Polak, P.; Haradhvala, N.; Lawrence, M.S.; Kiezun, A.; Fernandes, S.M.; Bahl, S.; et al. Whole-genome sequencing reveals activation-induced cytidine deaminase signatures during indolent chronic lymphocytic leukaemia evolution. Nat. Commun. 2015, 6, 8866. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, H.S.; Galashevskaya, A.; Doseth, B.; Sousa, M.M.; Sarno, A.; Visnes, T.; Aas, P.A.; Liabakk, N.-B.; Slupphaug, G.; Sætrom, P.; et al. AID expression in B-cell lymphomas causes accumulation of genomic uracil and a distinct AID mutational signature. DNA Repair 2015, 25, 60–71. [Google Scholar] [CrossRef]
- Patten, P.E.M.; Chu, C.C.; Albesiano, E.; Damle, R.N.; Yan, X.-J.; Kim, D.; Zhang, L.; Magli, A.R.; Barrientos, J.; Kolitz, J.E.; et al. IGHV-unmutated and IGHV-mutated chronic lymphocytic leukemia cells produce activation-induced deaminase protein with a full range of biologic functions. Blood 2012, 120, 4802–4811. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Sterling, J.; Thompson, C.; Harris, S.; Mav, D.; Shah, R.; Klimczak, L.J.; Kryukov, G.V.; Malc, E.; Mieczkowski, P.A.; et al. Clustered mutations in yeast and in human cancers can arise from damaged long single-strand DNA regions. Mol. Cell 2012, 46, 424–435. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Neumeister, P.; Goossens, T.; Nanjangud, G.; Chaganti, R.S.K.; Küppers, R.; Dalla-Favera, R. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature 2001, 412, 341–346. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Børresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [PubMed]
- Machado, H.E.; Mitchell, E.; Øbro, N.F.; Kübler, K.; Davies, M.; Leongamornlert, D.; Cull, A.; Maura, F.; Sanders, M.A.; Cagan, A.T.J.; et al. Diverse mutational landscapes in human lymphocytes. Nature 2022, 608, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Yamane, A.; Resch, W.; Kuo, N.; Kuchen, S.; Li, Z.; Sun, H.-W.; Robbiani, D.F.; McBride, K.; Nussenzweig, M.C.; Casellas, R. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat. Immunol. 2011, 12, 62–69. [Google Scholar] [CrossRef]
- Keim, C.; Kazadi, D.; Rothschild, G.; Basu, U. Regulation of AID, the B-cell genome mutator. Minerva Anestesiol. 2013, 27, 1–17. [Google Scholar] [CrossRef]
- Jiao, J.; Lv, Z.; Wang, Y.; Fan, L.; Yang, A. The off-target effects of AID in carcinogenesis. Front. Immunol. 2023, 14, 1221528. [Google Scholar] [CrossRef]
- Teater, M.; Dominguez, P.M.; Redmond, D.; Chen, Z.; Ennishi, D.; Scott, D.W.; Cimmino, L.; Ghione, P.; Chaudhuri, J.; Gascoyne, R.D.; et al. AICDA drives epigenetic heterogeneity and accelerates germinal center-derived lymphomagenesis. Nat. Commun. 2018, 9, 222. [Google Scholar] [CrossRef] [PubMed]
- Nonaka, T.; Toda, Y.; Hiai, H.; Uemura, M.; Nakamura, M.; Yamamoto, N.; Asato, R.; Hattori, Y.; Bessho, K.; Minato, N.; et al. Involvement of activation-induced cytidine deaminase in skin cancer development. J. Clin. Investig. 2016, 126, 1367–1382. [Google Scholar] [CrossRef] [PubMed]
- Qian, J.; Wang, Q.; Dose, M.; Pruett, N.; Kieffer-Kwon, K.-R.; Resch, W.; Liang, G.; Tang, Z.; Mathé, E.; Benner, C.; et al. B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell 2014, 159, 1524–1537. [Google Scholar] [CrossRef] [PubMed]
- Holmes, R.K.; Malim, M.H.; Bishop, K.N. APOBEC-mediated viral restriction: Not simply editing? Trends Biochem. Sci. 2007, 32, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy, M.; Patel, H.; Lai, C.-F.; Nguyen, V.T.; Nevedomskaya, E.; Harrod, A.; Russell, R.; Remenyi, J.; Ochocka, A.M.; Thomas, R.S.; et al. APOBEC3B-Mediated Cytidine Deamination Is Required for Estrogen Receptor Action in Breast Cancer. Cell Rep. 2015, 13, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Petljak, M.; Green, A.M.; Maciejowski, J.; Weitzman, M.D. Addressing the benefits of inhibiting APOBEC3-dependent mutagenesis in cancer. Nat. Genet. 2022, 54, 1599–1608. [Google Scholar] [CrossRef]
- Li, X.; Wang, Y.; Deng, S.; Zhu, G.; Wang, C.; Johnson, N.A.; Zhang, Z.; Tirado, C.R.; Xu, Y.; Metang, L.A.; et al. Loss of SYNCRIP unleashes APOBEC-driven mutagenesis, tumor heterogeneity, and AR-targeted therapy resistance in prostate cancer. Cancer Cell 2023, 41, 1427–1449.e12. [Google Scholar] [CrossRef] [PubMed]
- Isozaki, H.; Sakhtemani, R.; Abbasi, A.; Nikpour, N.; Stanzione, M.; Oh, S.; Langenbucher, A.; Monroe, S.; Su, W.; Cabanos, H.F.; et al. Therapy-induced APOBEC3A drives evolution of persistent cancer cells. Nature 2023, 620, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Klemm, L.; Duy, C.; Iacobucci, I.; Kuchen, S.; von Levetzow, G.; Feldhahn, N.; Henke, N.; Li, Z.; Hoffmann, T.K.; Kim, Y.-M.; et al. The B cell mutator AID promotes B lymphoid blast crisis and drug resistance in chronic myeloid leukemia. Cancer Cell 2009, 16, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Caswell, D.R.; Gui, P.; Mayekar, M.K.; Law, E.K.; Pich, O.; Bailey, C.; Boumelha, J.; Kerr, D.L.; Blakely, C.M.; Manabe, T.; et al. The role of APOBEC3B in lung tumor evolution and targeted cancer therapy resistance. Nat. Genet. 2023, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Garcia, N.M.G.; Becerra, J.N.; McKinney, B.J.; DiMarco, A.V.; Wu, F.; Fitzgibbon, M.; Alvarez, J.V. APOBEC3 activity promotes the survival and evolution of drug-tolerant persister cells during acquired resistance to EGFR inhibitors in lung cancer. bioRxiv 2023. [Google Scholar] [CrossRef]
- Petljak, M.; Alexandrov, L.B.; Brammeld, J.S.; Price, S.; Wedge, D.C.; Grossmann, S.; Dawson, K.J.; Ju, Y.S.; Iorio, F.; Tubio, J.M.; et al. Characterizing Mutational Signatures in Human Cancer Cell Lines Reveals Episodic APOBEC Mutagenesis. Cell 2019, 176, 1282–1294.e20. [Google Scholar] [CrossRef] [PubMed]
- Kingston, B.; Cutts, R.J.; Bye, H.; Beaney, M.; Walsh-Crestani, G.; Hrebien, S.; Swift, C.; Kilburn, L.S.; Kernaghan, S.; Moretti, L.; et al. Genomic profile of advanced breast cancer in circulating tumour DNA. Nat. Commun. 2021, 12, 2423. [Google Scholar] [CrossRef]
- Barroso-Sousa, R.; Jain, E.; Cohen, O.; Kim, D.; Buendia-Buendia, J.; Winer, E.; Lin, N.; Tolaney, S.; Wagle, N. Prevalence and mutational determinants of high tumor mutation burden in breast cancer. Ann. Oncol. 2020, 31, 387–394. [Google Scholar] [CrossRef] [PubMed]
- De Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014, 346, 251–256. [Google Scholar] [CrossRef] [PubMed]
- McGranahan, N.; Favero, F.; de Bruin, E.C.; Birkbak, N.J.; Szallasi, Z.; Swanton, C. Clonal status of actionable driver events and the timing of mutational processes in cancer evolution. Sci. Transl. Med. 2015, 7, 283ra54. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef] [PubMed]
- Roper, N.; Gao, S.; Maity, T.K.; Banday, A.R.; Zhang, X.; Venugopalan, A.; Cultraro, C.M.; Patidar, R.; Sindiri, S.; Brown, A.-L.; et al. APOBEC Mutagenesis and Copy-Number Alterations Are Drivers of Proteogenomic Tumor Evolution and Heterogeneity in Metastatic Thoracic Tumors. Cell Rep. 2019, 26, 2651–2666.e6. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.H.; Im, S.-A.; Park, K.; Wen, J.; Lee, K.-H.; Choi, Y.-L.; Lee, W.-C.; Min, A.; Bonato, V.; Park, S.; et al. Longitudinal multi-omics study of palbociclib resistance in HR-positive/HER2-negative metastatic breast cancer. Genome Med. 2023, 15, 55. [Google Scholar] [CrossRef]
- Law, E.K.; Sieuwerts, A.M.; LaPara, K.; Leonard, B.; Starrett, G.J.; Molan, A.M.; Temiz, N.A.; Vogel, R.I.; Gelder, M.E.M.-V.; Sweep, F.C.G.J.; et al. The DNA cytosine deaminase APOBEC3B promotes tamoxifen resistance in ER-positive breast cancer. Sci. Adv. 2016, 2, e1601737. [Google Scholar] [CrossRef] [PubMed]
- Durfee, C.; Temiz, N.A.; Levin-Klein, R.; Argyris, P.P.; Alsøe, L.; Carracedo, S.; de la Vega, A.A.; Proehl, J.; Holzhauer, A.M.; Seeman, Z.J.; et al. Human APOBEC3B promotes tumor development in vivo including signature mutations and metastases. Cell Rep. Med. 2023, 4, 101211. [Google Scholar] [CrossRef] [PubMed]
- Law, E.K.; Levin-Klein, R.; Jarvis, M.C.; Kim, H.; Argyris, P.P.; Carpenter, M.A.; Starrett, G.J.; Temiz, N.A.; Larson, L.K.; Durfee, C.; et al. APOBEC3A catalyzes mutation and drives carcinogenesis in vivo. J. Exp. Med. 2020, 217, e20200261. [Google Scholar] [CrossRef]
- Naumann, J.A.; Argyris, P.P.; Carpenter, M.A.; Gupta, H.B.; Chen, Y.; Temiz, N.A.; Zhou, Y.; Durfee, C.; Proehl, J.; Koniar, B.L.; et al. DNA Deamination Is Required for Human APOBEC3A-Driven Hepatocellular Carcinoma In Vivo. Int. J. Mol. Sci. 2023, 24, 9305. [Google Scholar] [CrossRef]
- Kidd, J.M.; Newman, T.L.; Tuzun, E.; Kaul, R.; E Eichler, E. Population stratification of a common APOBEC gene deletion polymorphism. PLoS Genet. 2007, 3, e63. [Google Scholar] [CrossRef]
- Rezaei, M.; Hashemi, M.; Hashemi, S.M.; Mashhadi, M.A.; Taheri, M. APOBEC3 Deletion is Associated with Breast Cancer Risk in a Sample of Southeast Iranian Population. Int. J. Mol. Cell. Med. 2015, 4, 103–108. [Google Scholar]
- Qi, G.; Xiong, H.; Zhou, C. APOBEC3 deletion polymorphism is associated with epithelial ovarian cancer risk among Chinese women. Tumor Biol. 2014, 35, 5723–5726. [Google Scholar] [CrossRef]
- Long, J.; Delahanty, R.J.; Li, G.; Gao, Y.-T.; Lu, W.; Cai, Q.; Xiang, Y.-B.; Li, C.; Ji, B.-T.; Zheng, Y.; et al. A common deletion in the APOBEC3 genes and breast cancer risk. J. Natl. Cancer Inst. 2013, 105, 573–579. [Google Scholar] [CrossRef]
- Wen, W.X.; Soo, J.S.-S.; Kwan, P.Y.; Hong, E.; Khang, T.F.; Mariapun, S.; Lee, C.S.-M.; Hasan, S.N.; Rajadurai, P.; Yip, C.H.; et al. Germline APOBEC3B deletion is associated with breast cancer risk in an Asian multi-ethnic cohort and with immune cell presentation. Breast Cancer Res. 2016, 18, 56. [Google Scholar] [CrossRef] [PubMed]
- Xuan, D.; Li, G.; Cai, Q.; Deming-Halverson, S.; Shrubsole, M.J.; Shu, X.-O.; Kelley, M.C.; Zheng, W.; Long, J. APOBEC3 deletion polymorphism is associated with breast cancer risk among women of European ancestry. Carcinogenesis 2013, 34, 2240–2243. [Google Scholar] [CrossRef]
- Göhler, S.; Filho, M.I.D.S.; Johansson, R.; Enquist-Olsson, K.; Henriksson, R.; Hemminki, K.; Lenner, P.; Försti, A. Impact of functional germline variants and a deletion polymorphism in APOBEC3A and APOBEC3B on breast cancer risk and survival in a Swedish study population. J. Cancer Res. Clin. Oncol. 2016, 142, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Gansmo, L.B.; Romundstad, P.; Hveem, K.; Vatten, L.; Nik-Zainal, S.; Lønning, P.E.; Knappskog, S. APOBEC3A/B deletion polymorphism and cancer risk. Carcinogenesis 2018, 39, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Gansmo, L.B.; Sofiyeva, N.; Bjørnslett, M.; Romundstad, P.; Hveem, K.; Vatten, L.; Dørum, A.; Lønning, P.E.; Knappskog, S. Impact of the APOBEC3A/B deletion polymorphism on risk of ovarian cancer. Sci. Rep. 2021, 11, 23463. [Google Scholar] [CrossRef]
- Caval, V.; Suspène, R.; Shapira, M.; Vartanian, J.-P.; Wain-Hobson, S. A prevalent cancer susceptibility APOBEC3A hybrid allele bearing APOBEC3B 3′UTR enhances chromosomal DNA damage. Nat. Commun. 2014, 5, 5129. [Google Scholar] [CrossRef]
- Nik-Zainal, S.; Wedge, D.C.; Alexandrov, L.B.; Petljak, M.; Butler, A.P.; Bolli, N.; Davies, H.R.; Knappskog, S.; Martin, S.; Papaemmanuil, E.; et al. Association of a germline copy number polymorphism of APOBEC3A and APOBEC3B with burden of putative APOBEC-dependent mutations in breast cancer. Nat. Genet. 2014, 46, 487–491. [Google Scholar] [CrossRef]
- Chan, K.; A Roberts, S.; Klimczak, L.J.; Sterling, J.F.; Saini, N.; Malc, E.P.; Kim, J.; Kwiatkowski, D.J.; Fargo, D.C.; A Mieczkowski, P.; et al. An APOBEC3A hypermutation signature is distinguishable from the signature of background mutagenesis by APOBEC3B in human cancers. Nat. Genet. 2015, 47, 1067–1072. [Google Scholar] [CrossRef]
- Middlebrooks, C.D.; Banday, A.R.; Matsuda, K.; Udquim, K.-I.; O Onabajo, O.; Paquin, A.; Figueroa, J.D.; Zhu, B.; Koutros, S.; Kubo, M.; et al. Association of germline variants in the APOBEC3 region with cancer risk and enrichment with APOBEC-signature mutations in tumors. Nat. Genet. 2016, 48, 1330–1338. [Google Scholar] [CrossRef]
- Rothman, N.; Garcia-Closas, M.; Chatterjee, N.; Malats, N.; Wu, X.F.; Figueroa, J.D.; Real, F.X.; Van den Berg, D.; Matullo, G.; Baris, D.; et al. A multi-stage genome-wide association study of bladder cancer identifies multiple susceptibility loci. Nat. Genet. 2010, 42, 978–984. [Google Scholar] [CrossRef]
- Matsuda, K.; Takahashi, A.; Middlebrooks, C.D.; Obara, W.; Nasu, Y.; Inoue, K.; Tamura, K.; Yamasaki, I.; Naya, Y.; Tanikawa, C.; et al. Genome-wide association study identified SNP on 15q24 associated with bladder cancer risk in Japanese population. Hum. Mol. Genet. 2015, 24, 1177–1184. [Google Scholar] [CrossRef]
- Mifsud, B.; Tavares-Cadete, F.; Young, A.N.; Sugar, R.; Schoenfelder, S.; Ferreira, L.; Wingett, S.W.; Andrews, S.; Grey, W.; A Ewels, P.; et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet. 2015, 47, 598–606. [Google Scholar] [CrossRef]
- Cannataro, V.L.; Mandell, J.D.; Townsend, J.P. Attribution of Cancer Origins to Endogenous, Exogenous, and Preventable Mutational Processes. Mol. Biol. Evol. 2022, 39, msac084. [Google Scholar] [CrossRef]
- Acha-Sagredo, A.; Ganguli, P.; Ciccarelli, F.D. Somatic variation in normal tissues: Friend or foe of cancer early detection? Ann. Oncol. 2022, 33, 1239–1249. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, K.; Gowers, K.H.C.; Lee-Six, H.; Chandrasekharan, D.P.; Coorens, T.; Maughan, E.F.; Beal, K.; Menzies, A.; Millar, F.R.; Anderson, E.; et al. Tobacco smoking and somatic mutations in human bronchial epithelium. Nature 2020, 578, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Di, L.; Li, J.; Fan, W.; Liu, Y.; Guo, W.; Liu, W.; Liu, L.; Li, Q.; Chen, L.; et al. A body map of somatic mutagenesis in morphologically normal human tissues. Nature 2021, 597, 398–403. [Google Scholar] [CrossRef]
- Moore, L.; Cagan, A.; Coorens, T.H.H.; Neville, M.D.C.; Sanghvi, R.; Sanders, M.A.; Oliver, T.R.W.; Leongamornlert, D.; Ellis, P.; Noorani, A.; et al. The mutational landscape of human somatic and germline cells. Nature 2021, 597, 381–386. [Google Scholar] [CrossRef]
- Lawson, A.R.J.; Abascal, F.; Coorens, T.H.H.; Hooks, Y.; O’neill, L.; Latimer, C.; Raine, K.; Sanders, M.A.; Warren, A.Y.; Mahbubani, K.T.A.; et al. Extensive heterogeneity in somatic mutation and selection in the human bladder. Science 2020, 370, 75–82. [Google Scholar] [CrossRef]
- Olafsson, S.; Rodriguez, E.; Lawson, A.R.J.; Abascal, F.; Huber, A.R.; Suembuel, M.; Jones, P.H.; Gerdes, S.; Martincorena, I.; Weidinger, S.; et al. Effects of psoriasis and psoralen exposure on the somatic mutation landscape of the skin. Nat. Genet. 2023, 55, 1892–1900. [Google Scholar] [CrossRef]
- Lee-Six, H.; Olafsson, S.; Ellis, P.; Osborne, R.J.; Sanders, M.A.; Moore, L.; Georgakopoulos, N.; Torrente, F.; Noorani, A.; Goddard, M.; et al. The landscape of somatic mutation in normal colorectal epithelial cells. Nature 2019, 574, 532–537. [Google Scholar] [CrossRef]
- Olafsson, S.; McIntyre, R.E.; Coorens, T.; Butler, T.; Jung, H.; Robinson, P.S.; Lee-Six, H.; Sanders, M.A.; Arestang, K.; Dawson, C.; et al. Somatic evolution in non-neoplastic IBD-affected colon. Cell 2020, 182, 672–684. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.C.; Robinson, P.S.; Coorens, T.H.; Yan, H.H.; Olafsson, S.; Lee-Six, H.; Sanders, M.A.; Siu, H.C.; Hewinson, J.; Yue, S.S.; et al. Mutational landscape of normal epithelial cells in Lynch Syndrome patients. Nat. Commun. 2022, 13, 2710. [Google Scholar] [CrossRef]
- Chang, J.; Zhao, X.; Wang, Y.; Liu, T.; Zhong, C.; Lao, Y.; Zhang, S.; Liao, H.; Bai, F.; Lin, D.; et al. Genomic alterations driving precancerous to cancerous lesions in esophageal cancer development. Cancer Cell 2023, 41, 2038–2050.e5. [Google Scholar] [CrossRef]
- Martincorena, I.; Fowler, J.C.; Wabik, A.; Lawson, A.R.; Abascal, F.; Hall, M.W.; Cagan, A.; Murai, K.; Mahbubani, K.; Stratton, M.R.; et al. Somatic mutant clones colonize the human esophagus with age. Science 2018, 362, 911–917. [Google Scholar] [CrossRef]
- Kakiuchi, N.; Yoshida, K.; Uchino, M.; Kihara, T.; Akaki, K.; Inoue, Y.; Kawada, K.; Nagayama, S.; Yokoyama, A.; Yamamoto, S.; et al. Frequent mutations that converge on the NFKBIZ pathway in ulcerative colitis. Nature 2020, 577, 260–265. [Google Scholar] [CrossRef]
- Robinson, P.S.; Thomas, L.E.; Abascal, F.; Jung, H.; Harvey, L.M.; West, H.D.; Olafsson, S.; Lee, B.C.; Coorens, T.H.; Lee-Six, H.; et al. Inherited MUTYH mutations cause elevated somatic mutation rates and distinctive mutational signatures in normal human cells. Nat. Commun. 2022, 13, 3949. [Google Scholar] [CrossRef]
- Robinson, P.S.; Coorens, T.H.; Palles, C.; Mitchell, E.; Abascal, F.; Olafsson, S.; Lee, B.C.; Lawson, A.R.; Lee-Six, H.; Moore, L.; et al. Increased somatic mutation burdens in normal human cells due to defective DNA polymerases. Nat. Genet. 2021, 53, 1434–1442. [Google Scholar] [CrossRef] [PubMed]
- Brunner, S.F.; Roberts, N.D.; Wylie, L.A.; Moore, L.; Aitken, S.J.; Davies, S.E.; Sanders, M.A.; Ellis, P.; Alder, C.; Hooks, Y.; et al. Somatic mutations and clonal dynamics in healthy and cirrhotic human liver. Nature 2019, 574, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.W.; Rouhani, F.J.; Brunner, S.F.; Brzozowska, N.; Aitken, S.J.; Yang, M.; Abascal, F.; Moore, L.; Nikitopoulou, E.; Chappell, L.; et al. Convergent somatic mutations in metabolism genes in chronic liver disease. Nature 2021, 598, 473–478. [Google Scholar] [CrossRef]
- Osorio, F.G.; Huber, A.R.; Oka, R.; Verheul, M.; Patel, S.H.; Hasaart, K.; de la Fonteijne, L.; Varela, I.; Camargo, F.D.; van Boxtel, R. Somatic Mutations Reveal Lineage Relationships and Age-Related Mutagenesis in Human Hematopoiesis. Cell Rep. 2018, 25, 2308–2316.e4. [Google Scholar] [CrossRef] [PubMed]
- Moore, L.; Leongamornlert, D.; Coorens, T.H.H.; Sanders, M.A.; Ellis, P.; Dentro, S.C.; Dawson, K.J.; Butler, T.; Rahbari, R.; Mitchell, T.J.; et al. The mutational landscape of normal human endometrial epithelium. Nature 2020, 580, 640–646. [Google Scholar] [CrossRef]
- Coorens, T.H.; Oliver, T.R.; Sanghvi, R.; Sovio, U.; Cook, E.; Vento-Tormo, R.; Haniffa, M.; Young, M.D.; Rahbari, R.; Sebire, N.; et al. Inherent mosaicism and extensive mutation of human placentas. Nature 2021, 592, 80–85. [Google Scholar] [CrossRef]
- Buhigas, C.; Warren, A.Y.; Leung, W.K.; Whitaker, H.C.; Luxton, H.J.; Hawkins, S.; Kay, J.; Butler, A.; Xu, Y.; Woodcock, D.J.; et al. The architecture of clonal expansions in morphologically normal tissue from cancerous and non-cancerous prostates. Mol. Cancer 2022, 21, 183. [Google Scholar] [CrossRef] [PubMed]
- Degasperi, A.; Zou, X.; Amarante, T.D.; Martinez-Martinez, A.; Koh, G.C.C.; Dias, J.M.L.; Heskin, L.; Chmelova, L.; Rinaldi, G.; Wang, V.Y.W.; et al. Substitution mutational signatures in whole-genome–sequenced cancers in the UK population. Science 2022, 376, abl9283. [Google Scholar] [CrossRef] [PubMed]
- Moody, S.; Senkin, S.; Islam, S.M.A.; Wang, J.; Nasrollahzadeh, D.; Penha, R.C.C.; Fitzgerald, S.; Bergstrom, E.N.; Atkins, J.; He, Y.; et al. Mutational signatures in esophageal squamous cell carcinoma from eight countries with varying incidence. Nat. Genet. 2021, 53, 1553–1563. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Chen, H.; Xi, R.; Cui, H.; Zhao, Y.; Xu, E.; Yan, T.; Lu, X.; Huang, F.; Kong, P.; et al. Whole-genome sequencing of 508 patients identifies key molecular features associated with poor prognosis in esophageal squamous cell carcinoma. Cell Res. 2020, 30, 902–913. [Google Scholar] [CrossRef] [PubMed]
- Thatikonda, V.; Islam, S.M.A.; Autry, R.J.; Jones, B.C.; Gröbner, S.N.; Warsow, G.; Hutter, B.; Huebschmann, D.; Fröhling, S.; Kool, M.; et al. Comprehensive analysis of mutational signatures reveals distinct patterns and molecular processes across 27 pediatric cancers. Nat. Cancer 2023, 4, 276–289. [Google Scholar] [CrossRef]
- Hänninen, U.A.; Katainen, R.; Tanskanen, T.; Plaketti, R.-M.; Laine, R.; Hamberg, J.; Ristimäki, A.; Pukkala, E.; Taipale, M.; Mecklin, J.-P.; et al. Exome-wide somatic mutation characterization of small bowel adenocarcinoma. PLoS Genet. 2018, 14, e1007200. [Google Scholar] [CrossRef]
- Henderson, S.; Chakravarthy, A.; Su, X.; Boshoff, C.; Fenton, T.R. APOBEC-mediated cytosine deamination links PIK3CA helical domain mutations to human papillomavirus-driven tumor development. Cell Rep. 2014, 7, 1833–1841. [Google Scholar] [CrossRef] [PubMed]
- Leshchiner, I.; Mroz, E.A.; Cha, J.; Rosebrock, D.; Spiro, O.; Bonilla-Velez, J.; Faquin, W.C.; Lefranc-Torres, A.; Lin, D.T.; Michaud, W.A.; et al. Inferring early genetic progression in cancers with unobtainable premalignant disease. Nat. Cancer 2023, 4, 550–563. [Google Scholar] [CrossRef]
- Yamanaka, S.; Balestra, M.E.; Ferrell, L.D.; Fan, J.; Arnold, K.S.; Taylor, S.; Taylor, J.M.; Innerarity, T.L. Apolipoprotein B mRNA-editing protein induces hepatocellular carcinoma and dysplasia in transgenic animals. Proc. Natl. Acad. Sci. USA 1995, 92, 8483–8487. [Google Scholar] [PubMed]
- Li, L.; Jiang, D.; Liu, H.; Guo, C.; Zhao, R.; Zhang, Q.; Xu, C.; Qin, Z.; Feng, J.; Liu, Y.; et al. Comprehensive proteogenomic characterization of early duodenal cancer reveals the carcinogenesis tracks of different subtypes. Nat. Commun. 2023, 14, 1751. [Google Scholar] [CrossRef] [PubMed]
- Jarmuza, A.; Chestera, A.; Baylissa, J.; Gisbournea, J.; Dunhamb, I.; Scottc, J.; Navaratnama, N. An anthropoid-specific locus of orphan C to U RNA-editing enzymes on chromosome 22. Genomics 2002, 79, 285–296. [Google Scholar] [CrossRef]
- Roberts, S.A.; Lawrence, M.S.; Klimczak, L.J.; A Grimm, S.A.; Fargo, D.; Stojanov, P.; Kiezun, A.; Kryukov, G.; Carter, S.L.; Saksena, G.; et al. An APOBEC cytidine deaminase mutagenesis pattern is widespread in human cancers. Nat. Genet. 2013, 45, 970–976. [Google Scholar] [CrossRef]
- Burns, M.B.; Lackey, L.; Carpenter, M.A.; Rathore, A.; Land, A.M.; Leonard, B.; Refsland, E.W.; Kotandeniya, D.; Tretyakova, N.; Nikas, J.B.; et al. APOBEC3B is an enzymatic source of mutation in breast cancer. Nature 2013, 494, 366–370. [Google Scholar] [CrossRef]
- Guo, H.; Zhu, L.; Huang, L.; Sun, Z.; Zhang, H.; Nong, B.; Xiong, Y. APOBEC Alteration Contributes to Tumor Growth and Immune Escape in Pan-Cancer. Cancers 2022, 14, 2827. [Google Scholar] [CrossRef]
- Lee, J.K.; Lee, J.; Kim, S.; Kim, S.; Youk, J.; Park, S.; An, Y.; Keam, B.; Kim, D.W.; Heo, D.S.; et al. Clonal History and Genetic Predictors of Transformation Into Small-Cell Carcinomas From Lung Adenocarcinomas. J. Clin. Oncol. 2017, 35, 3065–3074. [Google Scholar] [CrossRef]
- Boumelha, J.; Trécesson, S.d.C.; Law, E.K.; Romero-Clavijo, P.; Coelho, M.A.; Ng, K.W.; Mugarza, E.; Moore, C.; Rana, S.; Caswell, D.R.; et al. An Immunogenic Model of KRAS-Mutant Lung Cancer Enables Evaluation of Targeted Therapy and Immunotherapy Combinations. Cancer Res. 2022, 82, 3435–3448. [Google Scholar] [CrossRef] [PubMed]
- Alonso de la Vega, A.; Temiz, N.A.; Tasakis, R.; Somogyi, K.; Salgueiro, L.; Zimmer, E.; Ramos, M.; Diaz-Jimenez, A.; Chocarro, S.; Fernández-Vaquero, M.; et al. Acute expression of human APOBEC3B in mice results in RNA editing and lethality. Genome Biol. 2023, 24, 267. [Google Scholar]
Figure 1.
Existing lines of evidence implicating mutagenesis by APOBEC enzymes in cancer development and susceptibility.
Figure 1.
Existing lines of evidence implicating mutagenesis by APOBEC enzymes in cancer development and susceptibility.

Table 1.
Prevalence of reported APOBEC-mediated mutational signatures (SBS2 and SBS13) in available genomes from non-malignant tissues. In studies that do not discuss the prevalence of SBS2 and SBS13 (designated with “*”), any number of mutations designated as SBS2 or SBS13 in a sample were considered to indicate their presence. In some instances, low burdens of SBS2 and SBS13 may thus represent false-positive calls.
Table 1.
Prevalence of reported APOBEC-mediated mutational signatures (SBS2 and SBS13) in available genomes from non-malignant tissues. In studies that do not discuss the prevalence of SBS2 and SBS13 (designated with “*”), any number of mutations designated as SBS2 or SBS13 in a sample were considered to indicate their presence. In some instances, low burdens of SBS2 and SBS13 may thus represent false-positive calls.
| Study | Tissue Type | Percent of Subjects with SBS2 and/or SBS13 | Percent Samples with SBS2 and/or SBS13 |
|---|---|---|---|
| Yoshida et al., 2020 [77]. PMID: 31996850 | Lung: bronchus epithelium clones | 16/16 subjects | 493/632 (~78%) |
| Li et al., 2021 [78]. PMID: 34433965 | Lung: bronchus epithelium microbiopsies | 2/3 subjects | 15/135 (~11%) |
| Moore et al., 2021 [79]. PMID: 34433962 | Small intestine: epithelium crypts | 2/3 subjects | 36/49 (~73%) |
| Wang et al., 2023 [19]. PMID: 36702998 | Small intestine: epithelium crypts | 39/39 subjects | 58/342 (~17%) |
| Li et al., 2021 [78]. PMID: 34433965 | Small intestine: duodenum epithelium crypts | 2/4 subjects | 25/179 (~14%) |
| Lawson et al., 2020 [80]. PMID: 33004514 | Bladder: urothelium microbiopsies | 9/15 subjects | 19/88 (~22%) |
| Olafsson et al., 2023 [81]. PMID: 37884686 | Skin: epidermis microbiopsies | 12/111 subjects (patients with Psoriasis) * | 21/1182 (~2%) * |
| Lee-Six et al., 2019 [82]. PMID: 31645730 | Colon: epithelium crypts | 2/42 subjects | 2/445 (~0.5%) |
| Olafsson et al., 2020 [83]. PMID: 32697969 | Colon: epithelium crypts | 4/46 subjects (ulcerative colitis, n = 28; Crohn’s disease, n = 18) * | 26/446 (~6%) * |
| Lee et al., 2022 [84]. PMID: 35581206 | Intestine: epithelium crypts | 1/10 subjects (patients with Lynch syndrome) | 10/107 (~10%) |
| Li et al., 2021 [78]. PMID: 34433965 | Esophagus: epithelium microbiopsies | 1/5 subjects | 5/203 (~2%) |
| Chang et al., 2023 [85]. PMID: 38039962 | Esophagus: epithelium microbiopsies | 1/22 subjects | 1/48 (~2%) |
| Chang et al., 2023 [85]. PMID: 38039962 | Esophagus: low-grade intraepithelial neoplasia microbiopsies | 1/9 subjects | 1/23 (~4%) |
| Chang et al., 2023 [85]. PMID: 38039962 | Esophagus: high-grade intraepithelial neoplasia microbiopsies | 2/7 subjects | 2/8 (~28%) |
| Martincorena et al., 2018 [86]. PMID: 30337457 | Esophagus: epithelium microbiopsies | 0/21 subjects | 0/21 (0%) |
| Kakiuchi et al., 2020 [87]. PMID: 31853061 | Colon: epithelium crypts | 0/40 subjects (healthy, n = 22; ulcerative colitis, (n = 18) | 0/101 (0%) |
| Robinson et al., 2022 [88]. PMID: 35803914 | Intestine: epithelium crypts | 0/10 subjects (patients with BER deficiency) | 0/144 (0%) |
| Robinson et al., 2021 [89]. PMID: 34594041 | Intestine: epithelium crypts | 0/13 subjects (patients with POLE/POLD1 germline mutations) | 0/109 (0%) |
| Brunner et al., 2019 [90]. PMID: 31645727 | Liver: parenchyma microbiopsies | 0/14 subjects (healthy, n = 5; alcohol-related liver disease, n = 4; non-alcoholic fatty liver disease, n = 5) | 0/482 (0%) |
| Ng et al., 2021 [91]. PMID: 34646017 | Liver: parenchyma microbiopsies | 0/34 subjects (healthy, n = 5; alcohol-related liver disease, n = 10; non-alcoholic fatty liver disease, n = 19) | 0/1590 (0%) |
| Osorio et al., 2018 [92]. PMID: 30485801 | Bone marrow: clones (hematopoietic stem cells and multipotent progenitor cells) | 0/5 subjects | 0/18 (0%) |
| Machado et al., 2022 [32]. PMID: 35948631 | Blood: clones (native B, memory B, CD4+ and CD8+ native T cells, CD4+ and CD8+ memory T cells) | 0/7 subjects | 0/717 (0%) |
| Moore et al., 2020 [93]. PMID: 32350471 | Endometrium: gland microbiopsies | 28 subjects | 0/292 (0%) |
| Coorens et al., 2021 [94]. PMID: 33692543 | Placenta: bulk tissue | 0/37 subjects | 0/86 (0%) |
| Buhigas et al., 2022 [95]. PMID: 36131292 | Prostate: bulk tissue | 0/37 subjects | 0/51 (0%) |
| Li et al., 2021 [78]. PMID: 34433965 | Pan-tissue microbiopsies | Colon: 0/5 Gastric cardia: 0/3 Liver: 0/5 Pancreas: 0/5 Rectum: 0/4 Stomach: 0/3 | Colon: 0/246 (0%) Gastric cardia: 0/126 (0%) Liver: 0/248 (0%) Pancreas: 0/249 (0%) Rectum: 0/188 (0%) Stomach: 0/188 (0%) |
| Moore et al., 2021 [79]. PMID: 34433962 | Pan-tissue microbiopsies | Adrenal gland: 0/1 Appendix: 0/1 Bladder: 0/1 Bronchus: 0/1 Colon: 0/7 Heart: 0/1 Kidney: 0/2 Liver: 0/2 Esophagus: 0/2 Pancreas: 0/2 Prostate: 0/2 Skin: 0/2 Small bowel: 0/3 Stomach: 0/2 Testis: 0/13 Thyroid: 0/1 Ureter: 0/1 Visceral fat: 0/1 | Adrenal gland: 0/15 (0%) Appendix: 0/20 (0%) Bladder: 0/7 (0%) Bronchus: 0/22 (0%) Colon: 0/50 (0%) Heart: 0/6 (0%) Kidney: 0/19 (0%) Liver: 0/27 (0%) Esophagus: 0/30 (0%) Pancreas: 0/19 (0%) Prostate: 0/20 (0%) Skin: 0/14 (0%) Small bowel: 0/49 (0%) Stomach: 0/2 (0%) Testis: 0/209 (0%) Thyroid: 0/31 (0%) Ureter: 0/4 (0%) Visceral fat: 0/5 (0%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Dananberg, A.; Striepen, J.; Rozowsky, J.S.; Petljak, M. APOBEC Mutagenesis in Cancer Development and Susceptibility. Cancers 2024, 16, 374. https://doi.org/10.3390/cancers16020374
AMA Style
Dananberg A, Striepen J, Rozowsky JS, Petljak M. APOBEC Mutagenesis in Cancer Development and Susceptibility. Cancers. 2024; 16(2):374. https://doi.org/10.3390/cancers16020374
Chicago/Turabian StyleDananberg, Alexandra, Josefine Striepen, Jacob S. Rozowsky, and Mia Petljak. 2024. "APOBEC Mutagenesis in Cancer Development and Susceptibility" Cancers 16, no. 2: 374. https://doi.org/10.3390/cancers16020374
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.