


by
and

Epigenomes 2024, 8(1), 2; https://doi.org/10.3390/epigenomes8010002 - 27 Dec 2023
Abstract
Transposable elements (TEs) are major components of plant genomes with the ability to change their position in the genome or to create new copies of themselves in other positions in the genome. These can cause gene disruption and large-scale genomic alterations, including inversions,
[...] Read more.
Transposable elements (TEs) are major components of plant genomes with the ability to change their position in the genome or to create new copies of themselves in other positions in the genome. These can cause gene disruption and large-scale genomic alterations, including inversions, deletions, and duplications. Host organisms have evolved a set of mechanisms to suppress TE activity and counter the threat that they pose to genome integrity. These includes the epigenetic silencing of TEs mediated by a process of RNA-directed DNA methylation (RdDM). In most cases, the silencing machinery is very efficient for the vast majority of TEs. However, there are specific circumstances in which TEs can evade such silencing mechanisms, for example, a variety of biotic and abiotic stresses or in vitro culture. Hybridization is also proposed as an inductor of TE proliferation. In fact, the discoverer of the transposons, Barbara McClintock, first hypothesized that interspecific hybridization provides a “genomic shock” that inhibits the TE control mechanisms leading to the mobilization of TEs. However, the studies carried out on this topic have yielded diverse results, showing in some cases a total absence of mobilization or being limited to only some TE families. Here, we review the current knowledge about the impact of interspecific hybridization on TEs in plants and the possible implications of changes in the epigenetic mechanisms.
Full article
(This article belongs to the Collection Epigenetic Control in Plants)
►
Show Figures
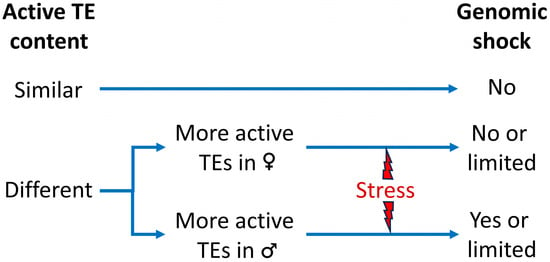
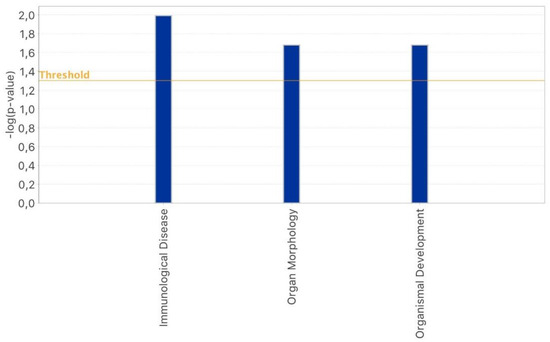
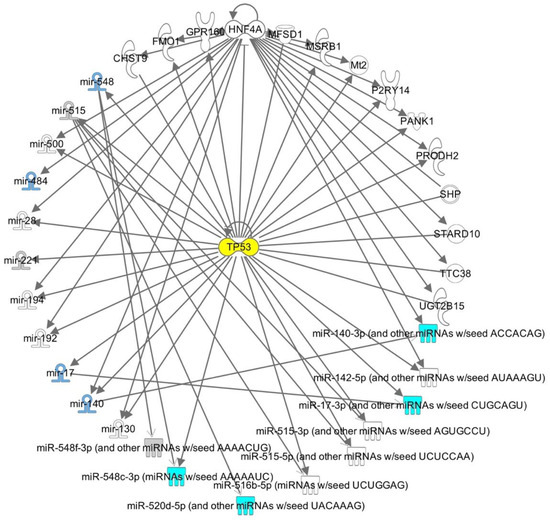
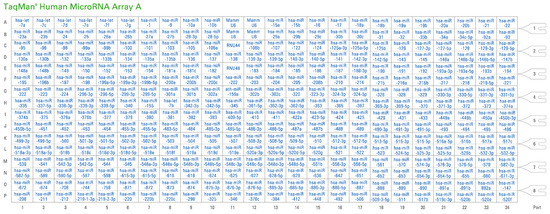
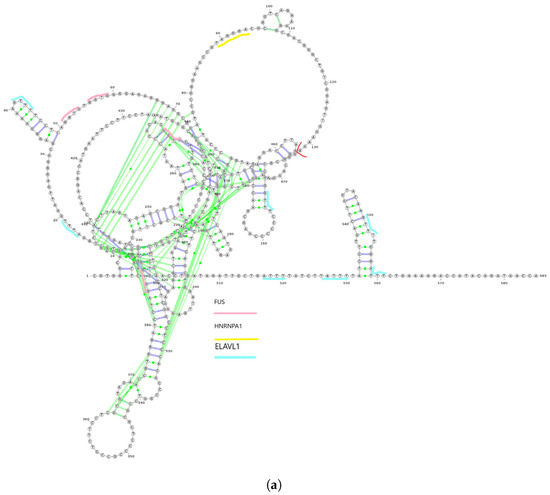
Figure 1














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}