






by
, , , , , , and

Genes 2024, 15(1), 102; https://doi.org/10.3390/genes15010102 - 15 Jan 2024
Abstract
Peanuts play a pivotal role as an economic crop on a global scale, serving as a primary source of both edible oil and protein. Peanut rust (Puccinia arachidis Speg.) disease constitutes a significant global biotic stress, representing a substantial economic threat to
[...] Read more.
Peanuts play a pivotal role as an economic crop on a global scale, serving as a primary source of both edible oil and protein. Peanut rust (Puccinia arachidis Speg.) disease constitutes a significant global biotic stress, representing a substantial economic threat to the peanut industry by inducing noteworthy reductions in seed yields and compromising oil quality. This comprehensive review delves into the distinctive characteristics and detrimental symptoms associated with peanut rust, scrutinizing its epidemiology and the control strategies that are currently implemented. Notably, host resistance emerges as the most favored strategy due to its potential to surmount the limitations inherent in other approaches. The review further considers the recent advancements in peanut rust resistance breeding, integrating the use of molecular marker technology and the identification of rust resistance genes. Our findings indicate that the ongoing refinement of control strategies, especially through the development and application of immune or highly resistant peanut varieties, will have a profound impact on the global peanut industry.
Full article
(This article belongs to the Special Issue 5Gs in Crop Genetic and Genomic Improvement)
►
Show Figures
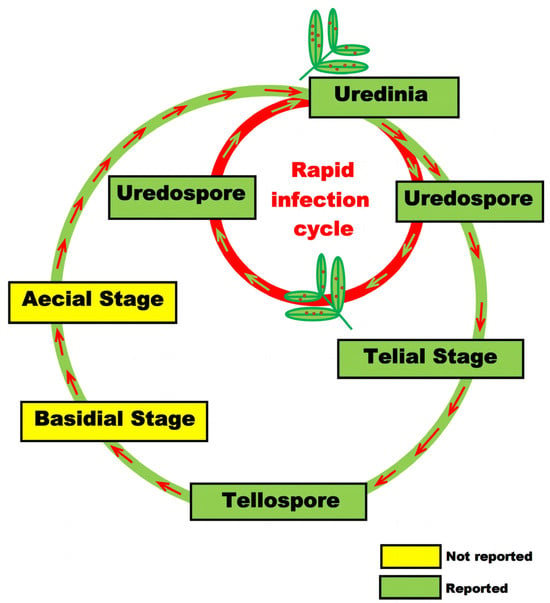
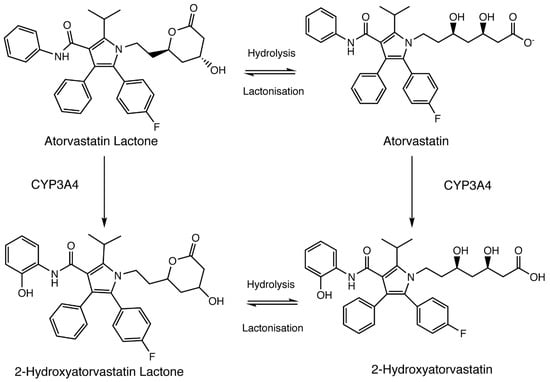
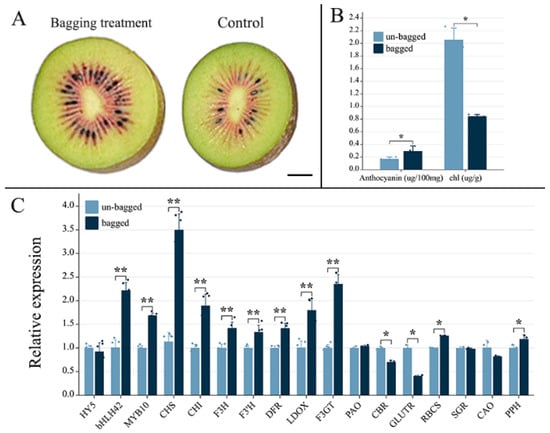
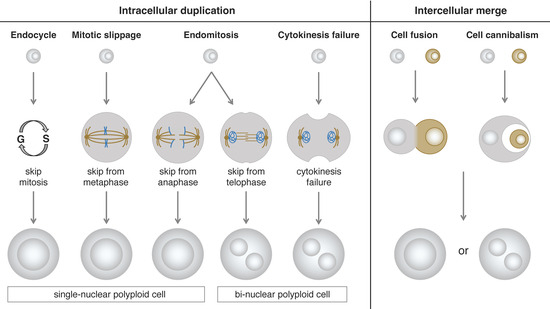
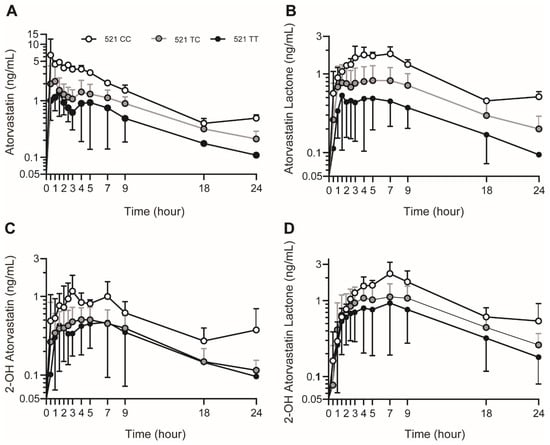
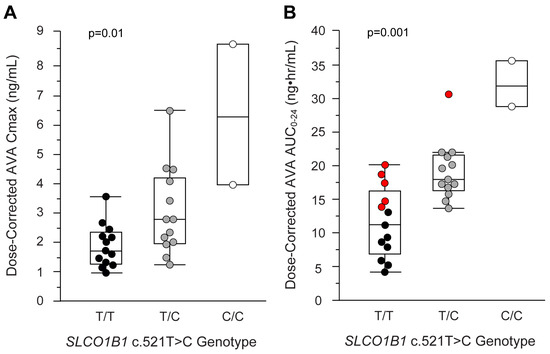
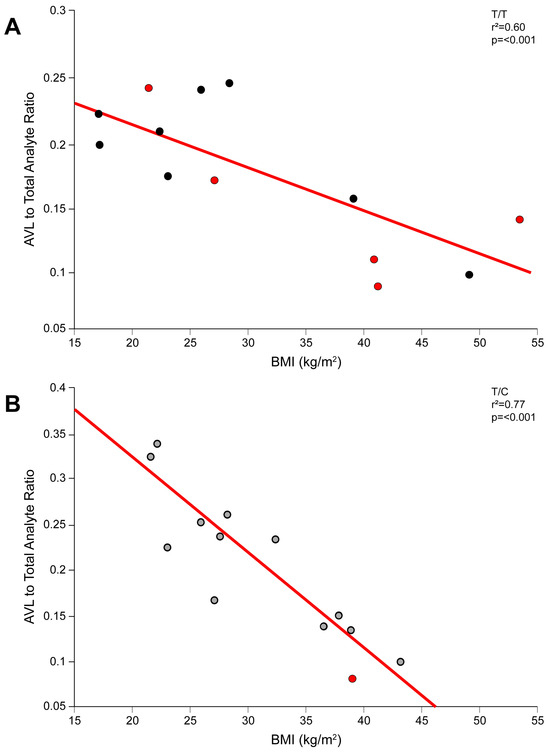
Figure 1











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
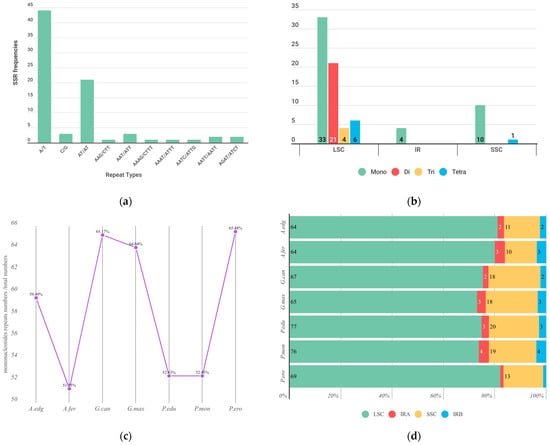{kind=link}
{kind=link}
{kind=link}
{kind=link}
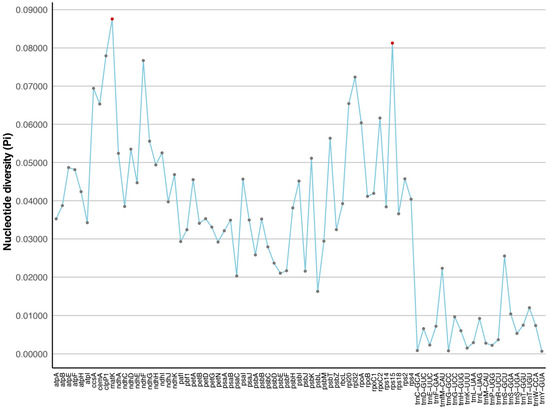{kind=link}
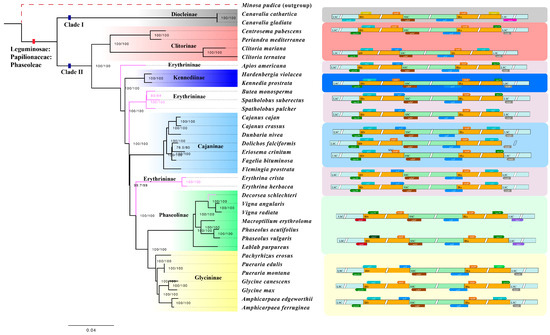{kind=link}
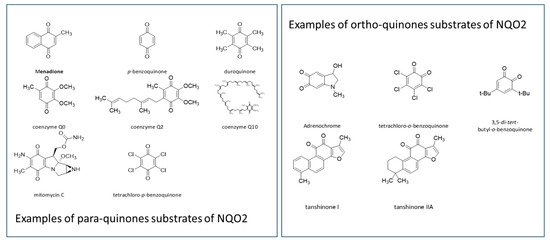{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}