


![Comprehensive Evaluation of End-Point Free Energy Techniques in Carboxylated-Pillar[6]arene Host-Guest Binding: IV. The QM Treatment, GB Models and the Multi-Trajectory Extension](https://pub.mdpi-res.com/title_story/title_story_17032268887149.jpg?1705329949)


Liquids 2024, 4(1), 107-116; https://doi.org/10.3390/liquids4010005 - 12 Jan 2024
Abstract
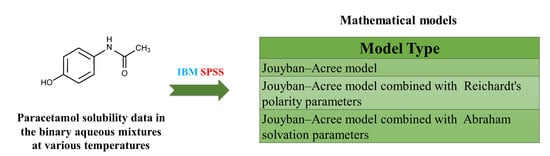
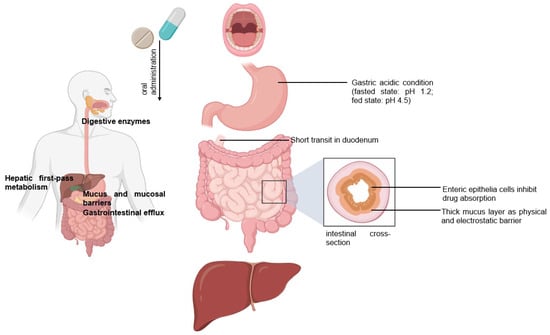
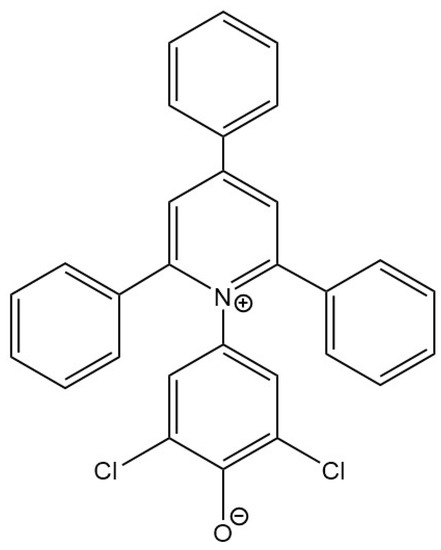
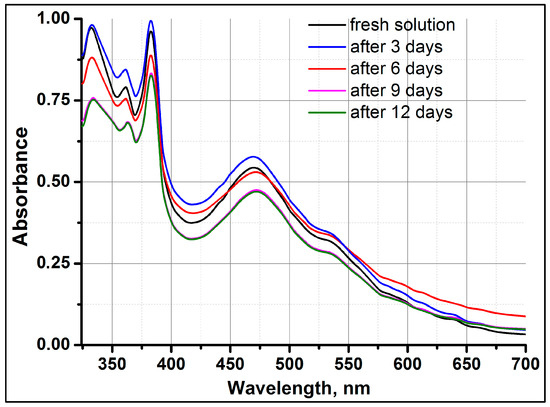
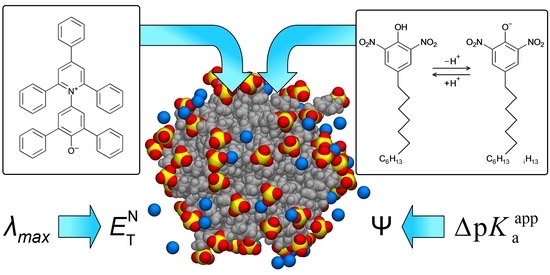
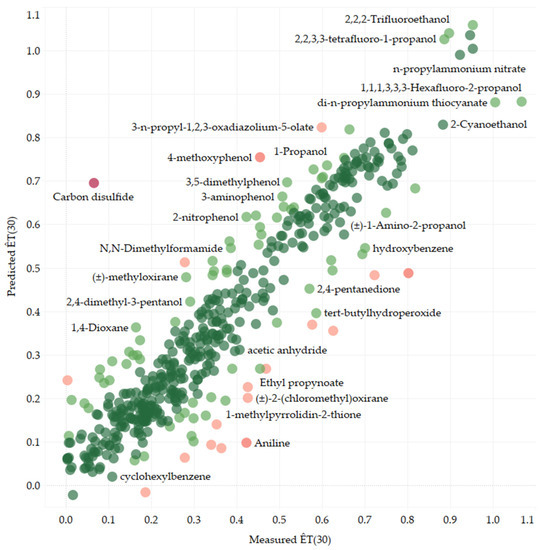
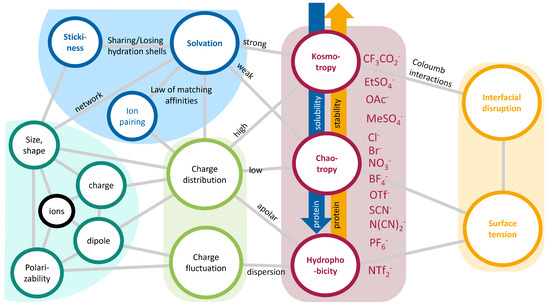
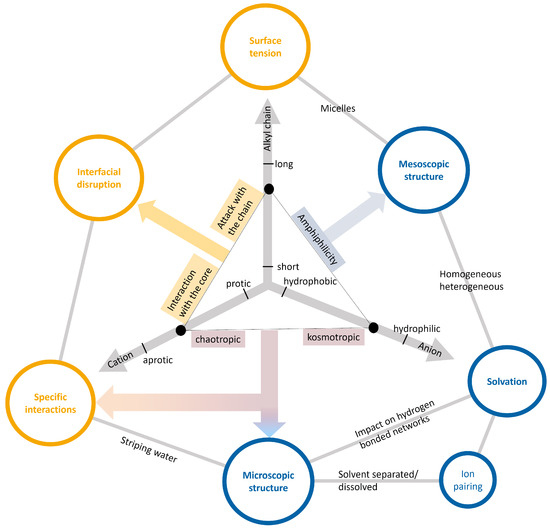
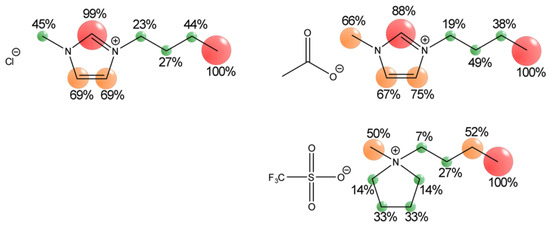
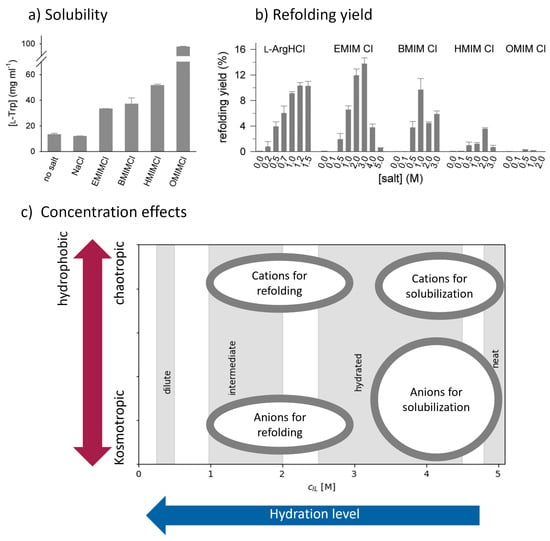
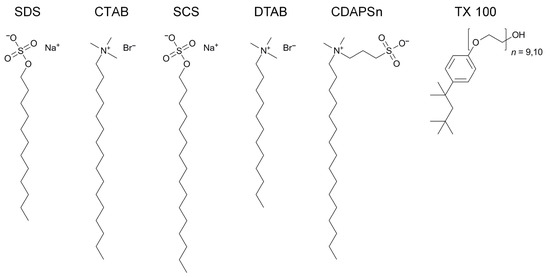
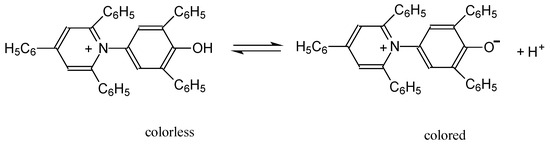
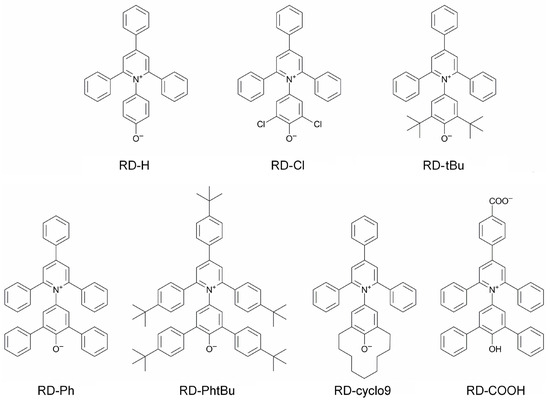
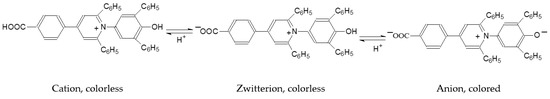
This short review describes the expansion of the solvatochromic approach utilizing water-soluble solvatochromic dyes to the analysis of solvent features of aqueous media in solutions of various compounds. These solvent features (polarity/dipolarity, hydrogen bond donor ability (HBD acidity), and hydrogen bond acceptor ability
[...] Read more.
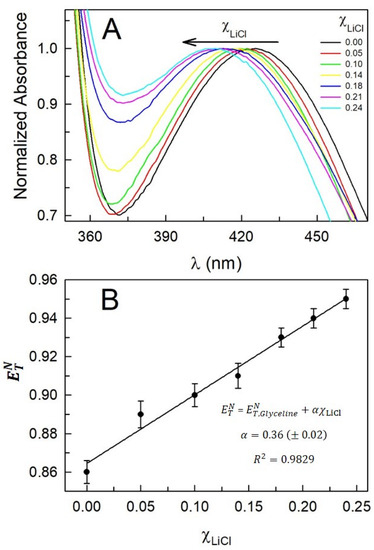
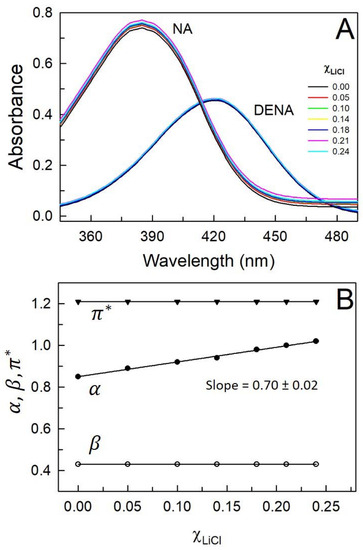
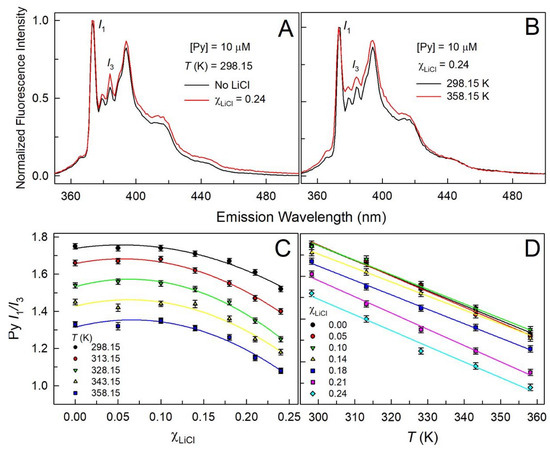
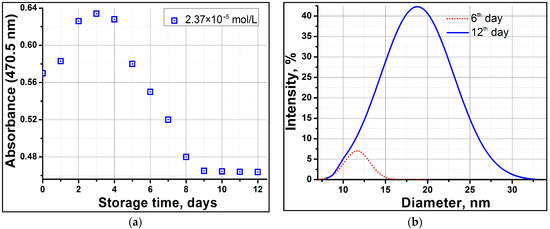
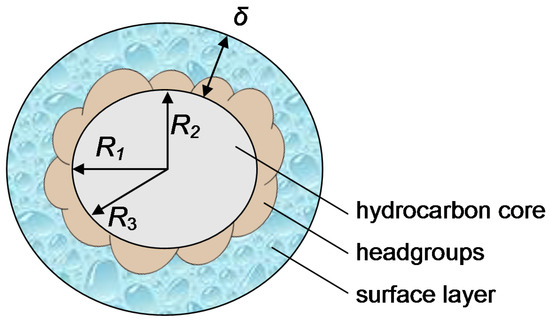
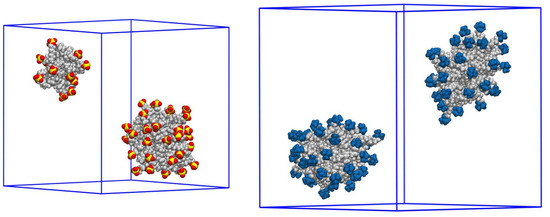
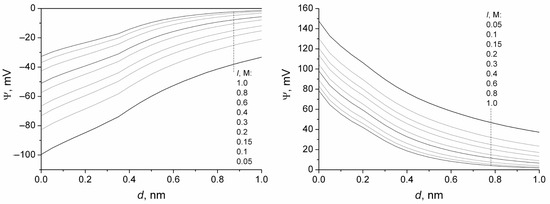
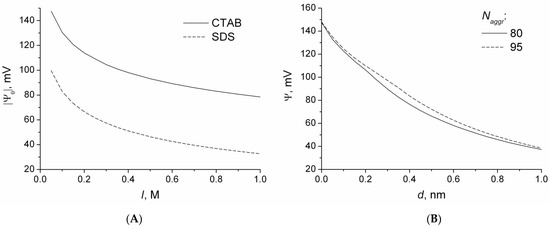
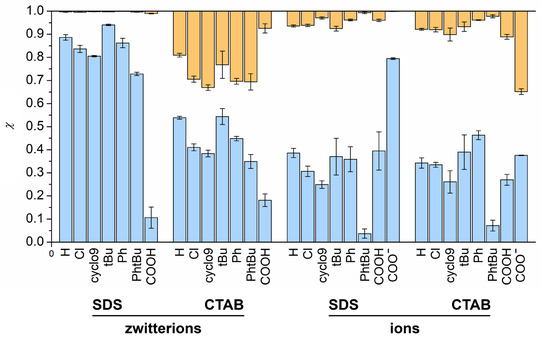
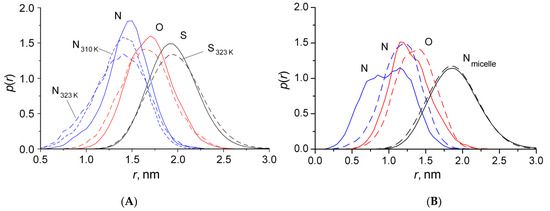
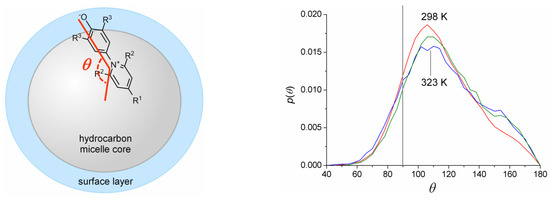
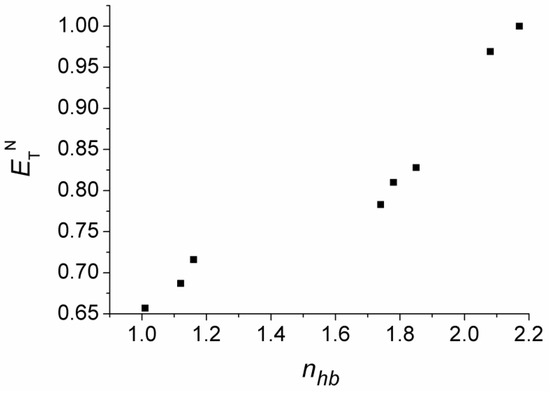
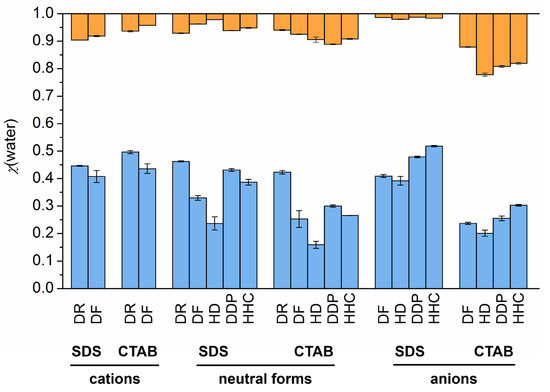
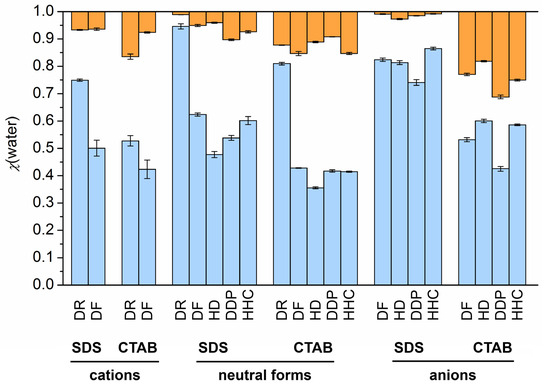
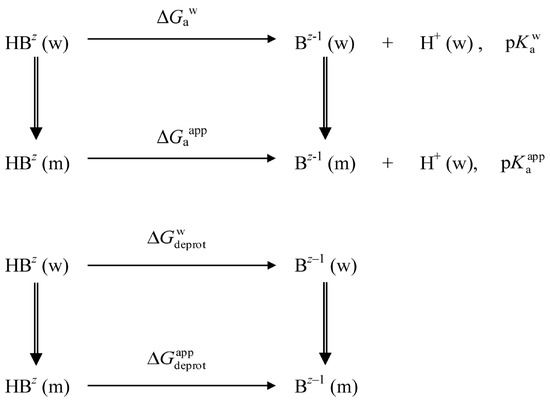
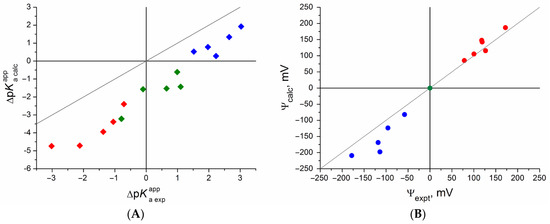
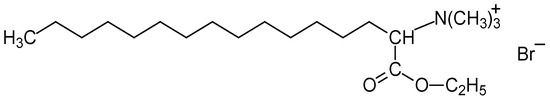
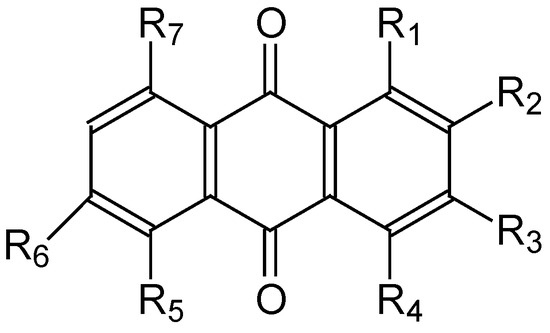
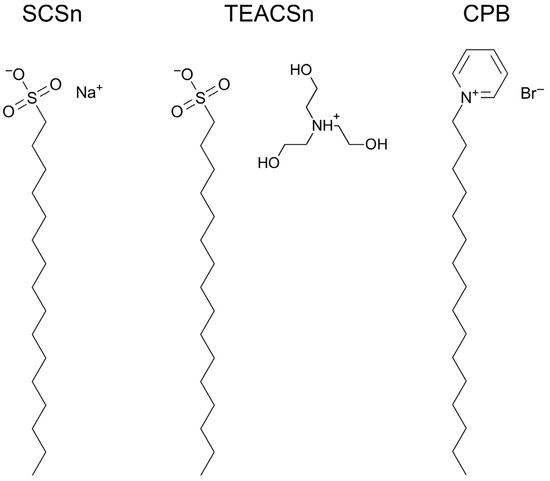
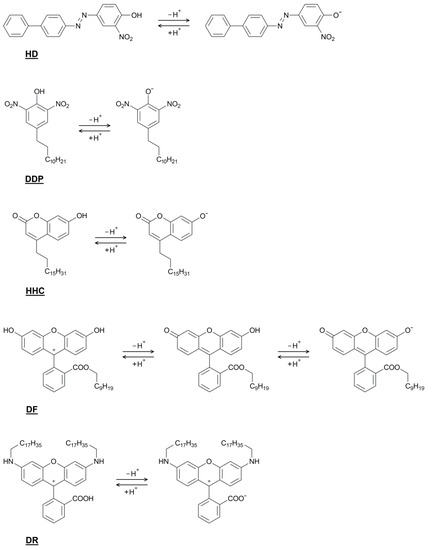
This short review describes the expansion of the solvatochromic approach utilizing water-soluble solvatochromic dyes to the analysis of solvent features of aqueous media in solutions of various compounds. These solvent features (polarity/dipolarity, hydrogen bond donor ability (HBD acidity), and hydrogen bond acceptor ability (HBA basicity)) vary depending on the nature and concentration of a solute. Furthermore, the solvent features of water (the solvent dipolarity/polarizability and hydrogen bond donor ability) in solutions of various compounds describe multiple physicochemical properties of these solutions (such as the solubility of various compounds in aqueous solutions, salting-out and salting-in constants for polar organic compounds in the presence of different inorganic salts, as well as water activity, osmotic coefficients, surface tension, viscosity, and the relative permittivity of aqueous solutions of different individual compounds) and are likely related to changes in the arrangement of hydrogen bonds of water in these solutions.
Full article
(This article belongs to the Special Issue Solvatochromic Probes and Their Applications in Molecular Interaction Studies—a Themed Issue to Honor Professor Dr. Christian Reichardt)
►
Show Figures
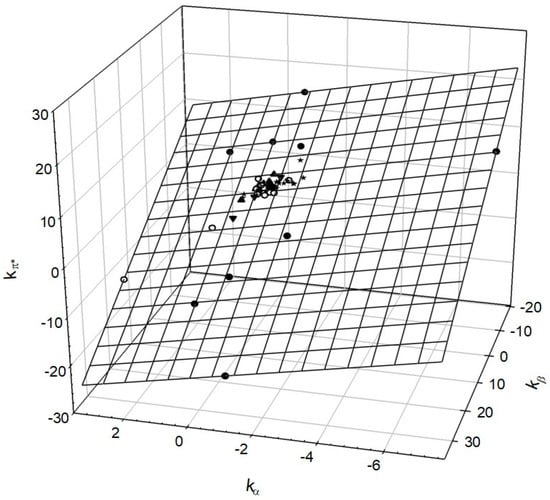
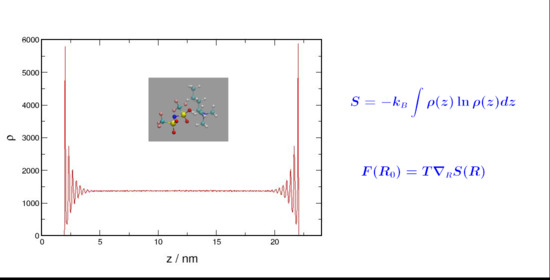
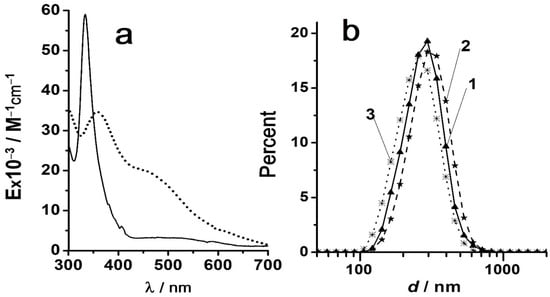
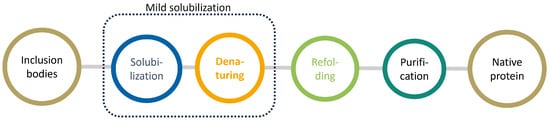
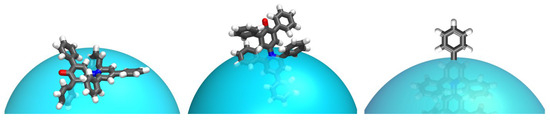
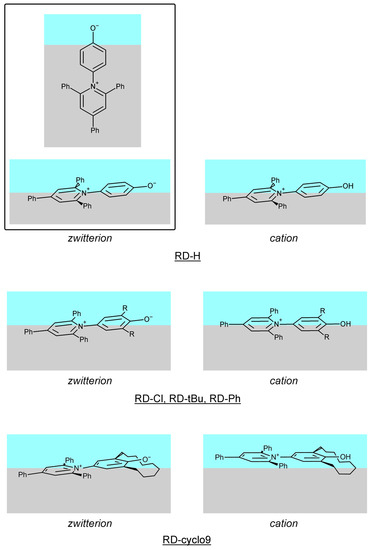
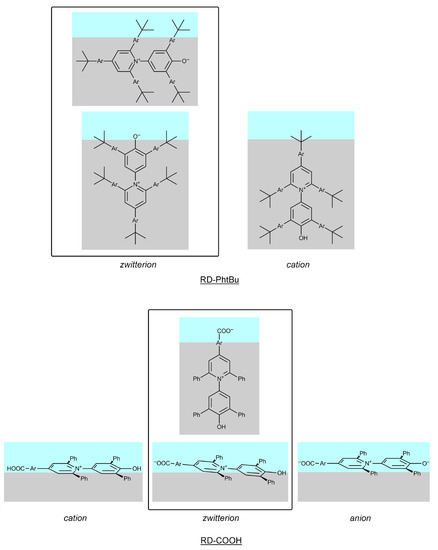
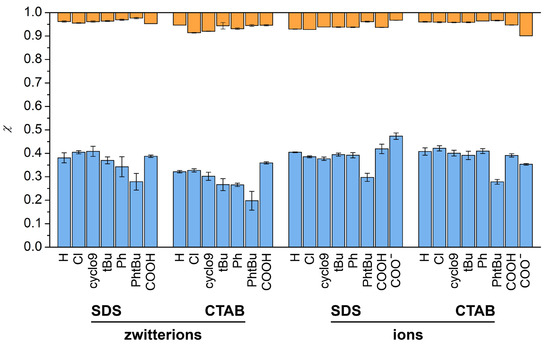
Figure 1














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}