





Pathogens 2024, 13(1), 78; https://doi.org/10.3390/pathogens13010078 - 15 Jan 2024
Abstract
The pathogenic mycoplasmas are among the bacteria causing significant losses in the poultry industry worldwide. Mycoplasma gallisepticum (MG) and M. synoviae (MS) are economically important pathogens causing chronic respiratory disease, decreased growth, egg production and hatchability rates, and significant downgrading of carcasses. Effective
[...] Read more.
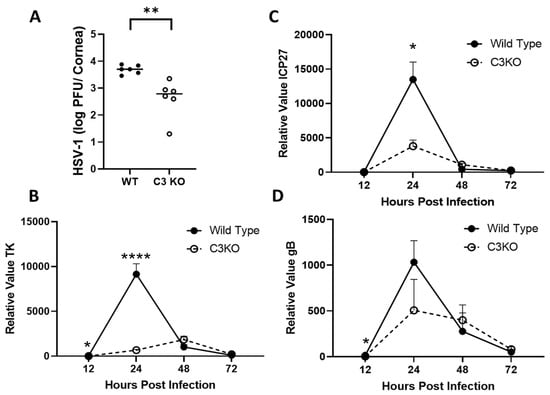
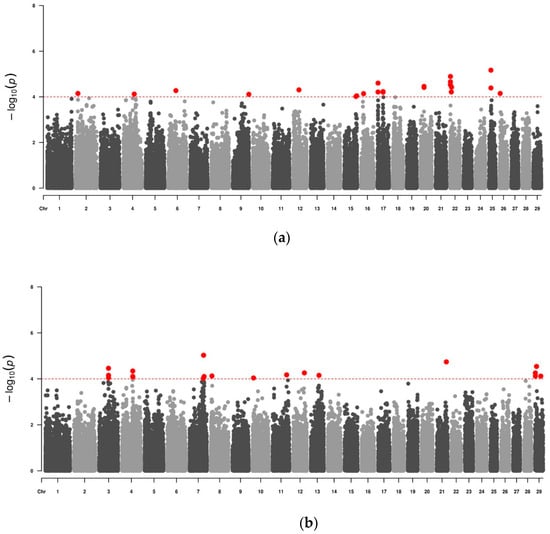
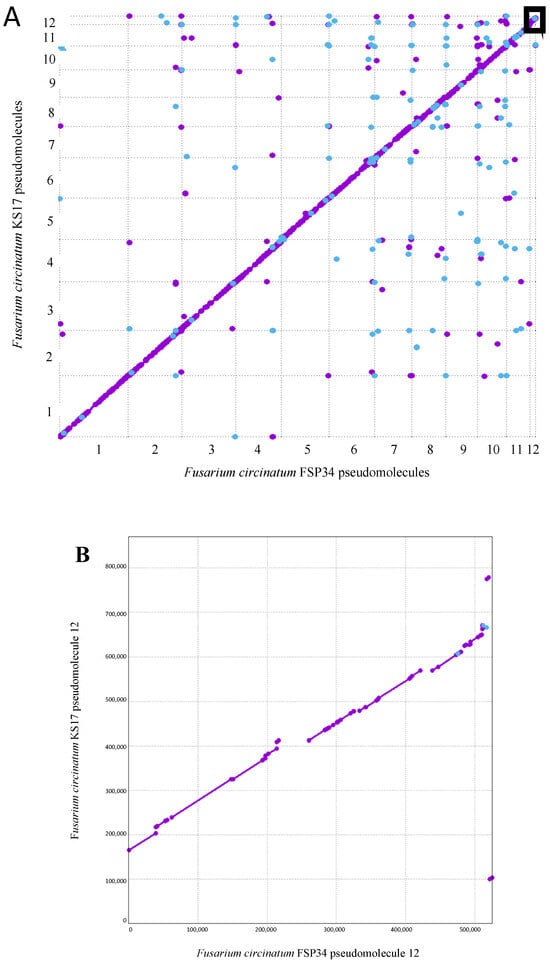
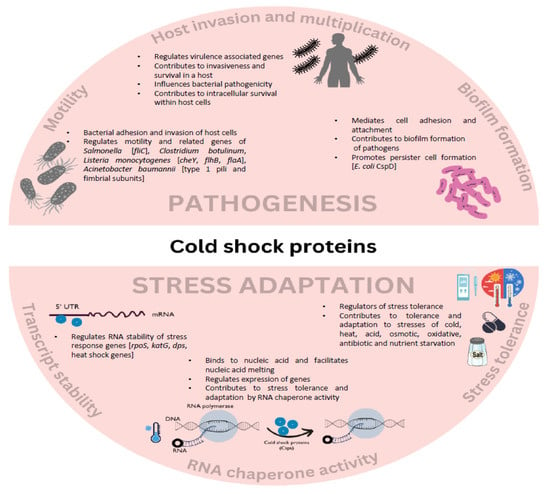
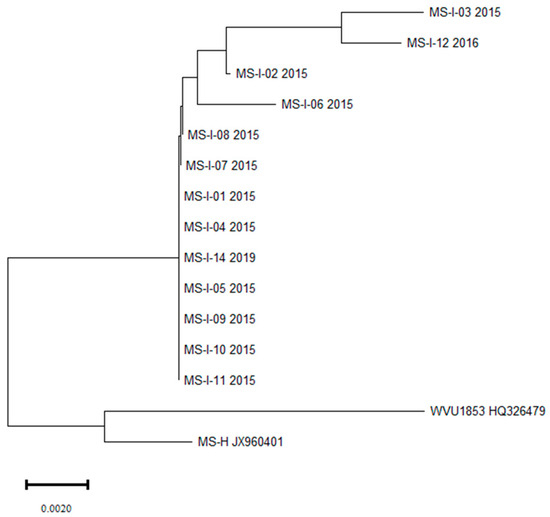
The pathogenic mycoplasmas are among the bacteria causing significant losses in the poultry industry worldwide. Mycoplasma gallisepticum (MG) and M. synoviae (MS) are economically important pathogens causing chronic respiratory disease, decreased growth, egg production and hatchability rates, and significant downgrading of carcasses. Effective diagnosis of infection with these species in poultry is highly requisite considering their two routes of spreading—horizontal and vertical. Their prevalence and molecular epidemiology were investigated in 184 turkey flocks in Poland. Tracheal samples were selected from 144 broiler flocks and 40 turkey breeder flocks collected in 2015–2023. The prevalence of MG was determined by real-time PCR targeting the 16S rRNA gene and PCR targeting the mgc2 gene, and MS was determined by a 16–23S rRNA real-time PCR and a vlhA gene PCR. Further identification and molecular characterization were carried out using PCR and sequencing. M. gallisepticum and M. synoviae were found in 8.33% and 9.72% of turkey broiler flocks respectively. The phylogenetic analysis of MG isolates in most cases showed high similarity to the ts-11-like strains. MS isolates showed high similarity to strains isolated from flocks of laying hens causing EAA. Additional tests detected Ornithobacterium rhinotracheale, Gallibacterium anatis, Enterococcus faecalis and Enterococcus faecium, Staphylococcus aureus and Riemerella anatipestifer. These secondary pathogens could have significantly heightened the pathogenicity of the mycoplasma infections studied.
Full article
(This article belongs to the Section Bacterial Pathogens)
►
Show Figures
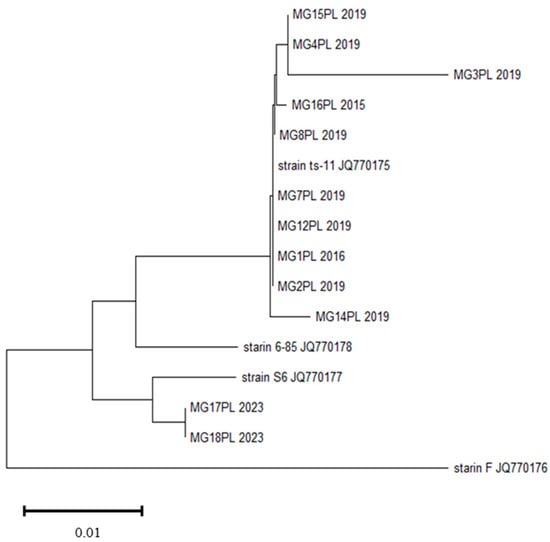
Figure 1





.jpg)







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}